


Krzysztof Brzozowski
Wydział Chemii, Uniwersytet Gdański
strony wersji drukowanej: 18-27

strony wersji drukowanej: 18-27
Nowoczesne metody spektroskopowe zrewolucjonizowały proces poznawania i identyfikacji biopolimerów. Początkowo z widm magnetycznego rezonansu jądrowego można było odczytać jedynie informacje na temat połączeń atomów oraz podstawowej stereochemii. Duża zawartość atomów wodoru w cząsteczkach organicznych spowodowała, że są one traktowane jak wewnętrzne sondy, a spektroskopia 1H MRJ stała się techniką fundamentalną wśród metod spektroskopowych. Wprowadzenie technik impulsowych w połączeniu z transformacją Fouriera pozwoliło na rejestrowanie widm dla małej ilości substancji i skrócenie całkowitego czasu analizy. Pozwoliło to również na rutynowe rejestrowanie widm węglowych, co było wcześniej niemożliwe ze względu na małązawartość izotopu 13C (około 1.11%) w badanych próbkach. Kolejnym ważnym krokiem w rozwoju spektroskopii MRJ było wprowadzenie eksperymentów wielowymiarowych. Dostarczają one cennych informacji na temat dipolowych i skalarnych sprzężeń pomiędzy jądrami atomowymi.
Udoskonalenia i rozwój magnesów nadprzewodzących i elektroniki sterującej stopniowo poprawiał proces rejestracji widm. Znaczące zmiany dały się zauważyć w momencie wprowadzenia gradientowych metod pulsowych (PFG- ang. Pulse Field Gradient) a później kriosond. Dzięki zastosowaniu PFG wpływ warunków zewnętrznych i fluktuacje pola magnetycznego są słabiej widoczne na rejestrowanych widmach. Dodatkowym atutemwynikającym z zastosowania kriosondy jest duży stosunek sygnału do szumu, który jest bardziej korzystny wraz z obniżaniem temperatury pomiaru. Chłodzone ciekłym helem kriosondy pozwalają na rejestrację widm w zakresie temperatur od 233 do 353 K.
Alternatywą dla krystalografii rentgenowskiej – szeroko stosowanej w badaniu białek jest CP MAS (ang. Cross-Polarization Magic Angle Spinning) MRJ ciała stałego. Technika ta podobnie jak w przypadku użycia kriosondy pozwala na rejestrowanie widm z małej ilości substancji jednocześnie eliminując problem agregacji.
Spektroskopia MRJ i krystalografia rentgenowska należą do najbardziej dokładnych metod analizy struktury i konformacji. Tak naprawdę w przypadku badania biopolimerów są technikami komplementarnymi. Obie pozwalają na badanie układów na poziomie atomowym. Oczywistym jest, że obie te techniki posiadają zarówno wady jak i zalety. Największą przewagą MRJ nad krystalografią jest możliwość wykonywania pomiarów w roztworze, który często jest naturalnym środowiskiem występowania i działania większości biopolimerów. Z drugiej jednak strony wielkość badanej cząsteczki stanowi poważne ograniczenie. Białko o masie powyżej 30 kDa może być problemem dla spektroskopii MRJ w przeciwieństwie do krystalografii rentgenowskiej.
Pomimo faktu, że spektroskopia MRJ jest wartościowym źródłem informacji strukturalnych powinna być wykorzystywana z innymi dodatkowymi metodami badawczymi. Takie połączenie pozwoli dokładnie wytłumaczyć zależność struktura-aktywność.
Jak to działa?
W spektroskopii MRJ wykorzystuje się magnetyczne właściwości jąder atomowych izotopów, w przypadku których magnetyczna liczba spinowa jest różna od 0. Kiedy jądro atomowe znajdzie się polu magnetycznym, jego wektor magnetyzacji jest odchylany o kąt 90o pod wpływem impulsu o częstości radiowej - RF. Po wzbudzeniu wszystkie wektory powracają do stanu równowagi emitując pewną porcję energii – rys. 1.
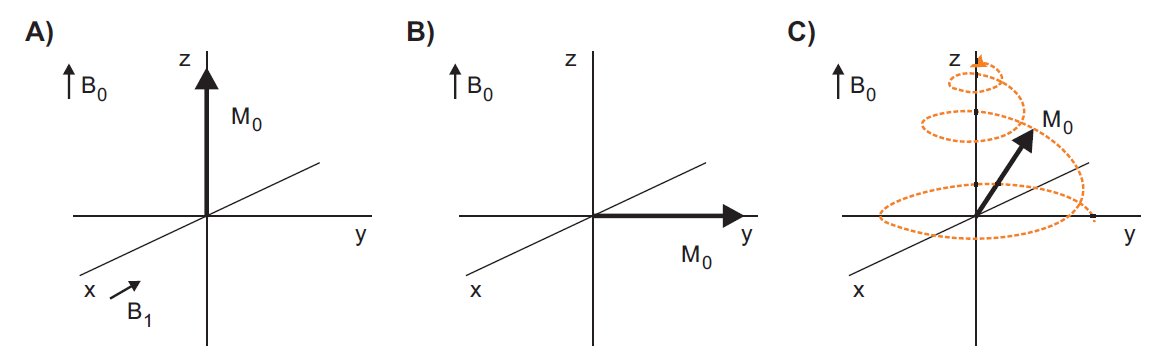
Rysunek 1. Zachowanie wektora magnetyzacji próbki: A) po umieszczeniu w polu magnetycznym, B) po działaniu pulsem o częstości radiowej B1, C) zanik precesji (relaksacja) wektora magnetycznego próbki- linia przerywana.
Detektor rejestruje zmiany energetyczne podczas tzw. zaniku swobodnej precesji wektora magnetycznego jądra. Zarejestrowany w domenie czasu sygnał FID (ang. Free Induction Decay) zawiera informacje na temat wszystkich wzbudzonych jąder atomowych obecnych w cząsteczce. Następnie te informacje konwertowane są w widmo w domenie częstotliwości za pomocą transformacji Fouriera – rys. 2.
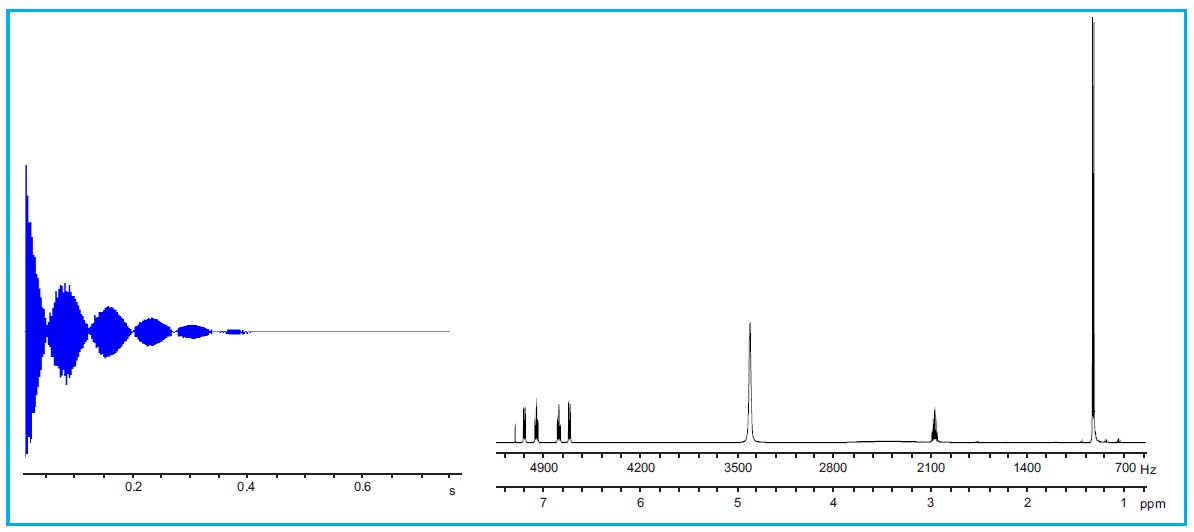
Rysunek 2. Przykładowy sygnał FID (po lewej) i widmo MRJ po transformacji Fouriera (po prawej).
Warunkiem koniecznym do uzyskania dobrego widma MRJ (o korzystnym stosunku sygnału do szumu) jest poprawne wyznaczenie tak zwanego pulsu 90o. Polega to na pomiarze serii widm dla sygnału referencyjnego działając na próbkę różnymi pulsami RF. Najbardziej intensywny sygnał odpowiada poszukiwanemu pulsowi 90o - rys. 3. Poprawne wyznaczenie tego pulsu jest szczególnie ważne w przypadku rejestrowania widm dla biopolimerów lub izolowanego materiału biologicznego, ponieważ bardzo często stężenie badanej próbki jest małe.
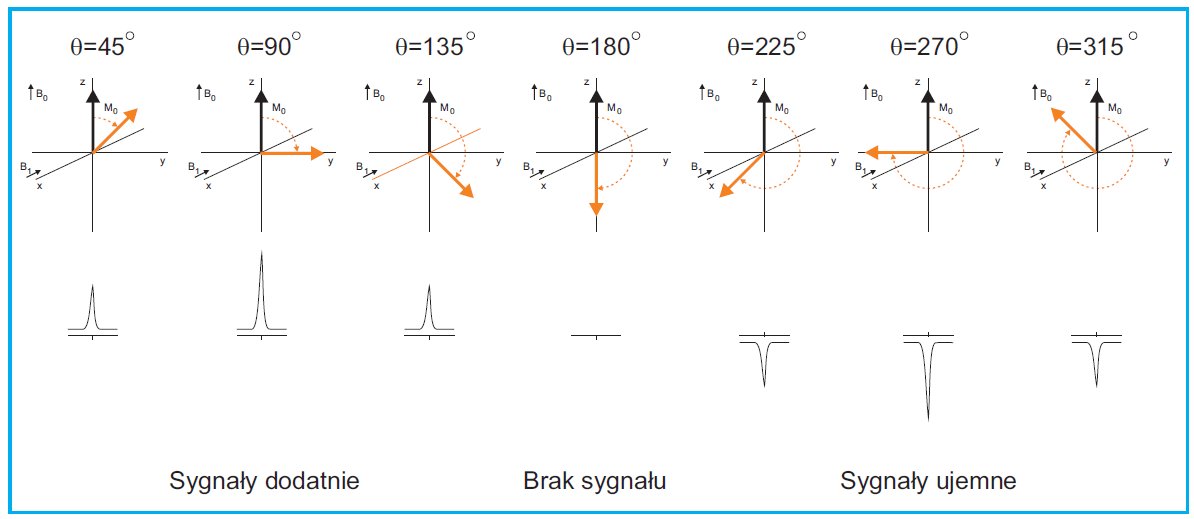
Rysunek 3. Wpływ pulsu RF na wektor magnetyzacji próbki i intensywność sygnału.
Wykorzystywany sprzęt
Kluczowym elementem spektrometru MRJ jest magnes. Z uwagi na fakt, że biomolekuły, w tym również peptydy i białka, zawierają dużo protonów ich częstotliwość rezonansowa jest podstawowym parametrem opisującym częstotliwość podstawową spektrometru. Obecnie magnesy nadprzewodzące wykorzystuje się do rutynowych jak i bardzo wyrafinowanych analiz. Wysokie pole (nawet około 1 GHz), zastosowanie kriosond i transformacji Fouriera pozwala na przeprowadzanie badań dla próbek biologicznych o niewielkim stężeniu. Na rysunku 4 pokazano schemat budowy magnesu.
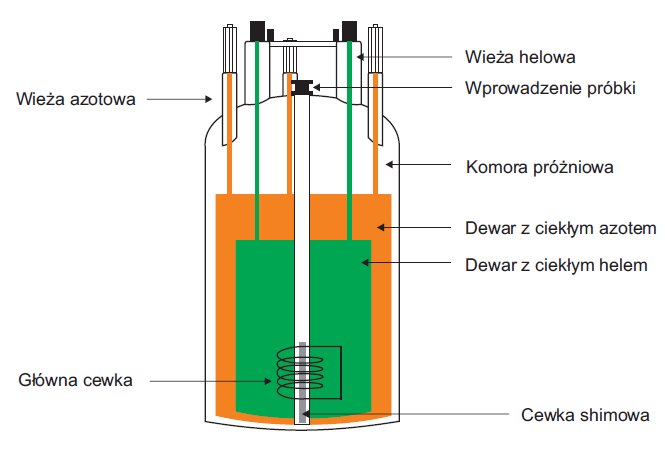
Rysunek 4. Schemat magnesu nadprzewodzącego.
Aby zagwarantować długą i bezawaryjną pracę całego systemu ważne jest aby zapewnić odpowiednie chłodzenie. Główny element magnesu - zrobiona z materiału nadprzewodzącego cewka zanurzona jest w ciekłym helu. W kolejnym zbiorniku znajduje się ciekły azot, którego zadaniem jest zmniejszenie odparowania ciekłego helu. Całość jest izolowana „płaszczem próżniowym”. Dodatkową rzeczą, o którą należy zadbać to wyeliminowanie drgań podłoża.
Sam magnes jednak to nie wszystko. Wiele zależy od zastosowanej sondy pomiarowej. Wyróżnić można cztery podstawowe typy tego urządzenia:
- dwukanałowa sonda szerokopasmowa – jest ona zoptymalizowana do pomiaru widm z tzw. odsprzęganiem od protonów. Pozwala na rejestrację widm 1D dla jąder z zakresu 31P-15N i oczywiście 13C,
- dwukanałowa inwersyjna sonda szerokopasmowa – również pozwala na rejestrację sygnałów jąder z zakresu 31P-15N, jednak jest ona dedykowana dla dwuwymiarowych eksperymentów heterokorelacyjnych (omówionych później) HSQC, HMQC czy HMBC,
- trójkanałowa sonda – jest powszechnie stosowana do badania biomolekuł, w tym biopolimerów. Jest nieoceniona podczas badań konformacyjnych ponieważ pozwala na pomiar sygnałów pochodzących jednocześnie od trzech jąder. Pozwala na rejestrację widm wielowymiarowych,
- kriosonda – chłodzona ciekłym helem, czterokanałowa sonda do badań strukturalnych biomolekuł. Cztery kanały pozwalają na jednoczesny pomiar jąder izotopów: 1H, 13C, 31P, 15N. Temperatura pracy sondy to około 20 K co powoduje 400% wzrost czułości w porównaniu z sondami szerokopasmowymi.
Podstawowe widma wykorzystywane do ustalenia struktury peptydu lub białka
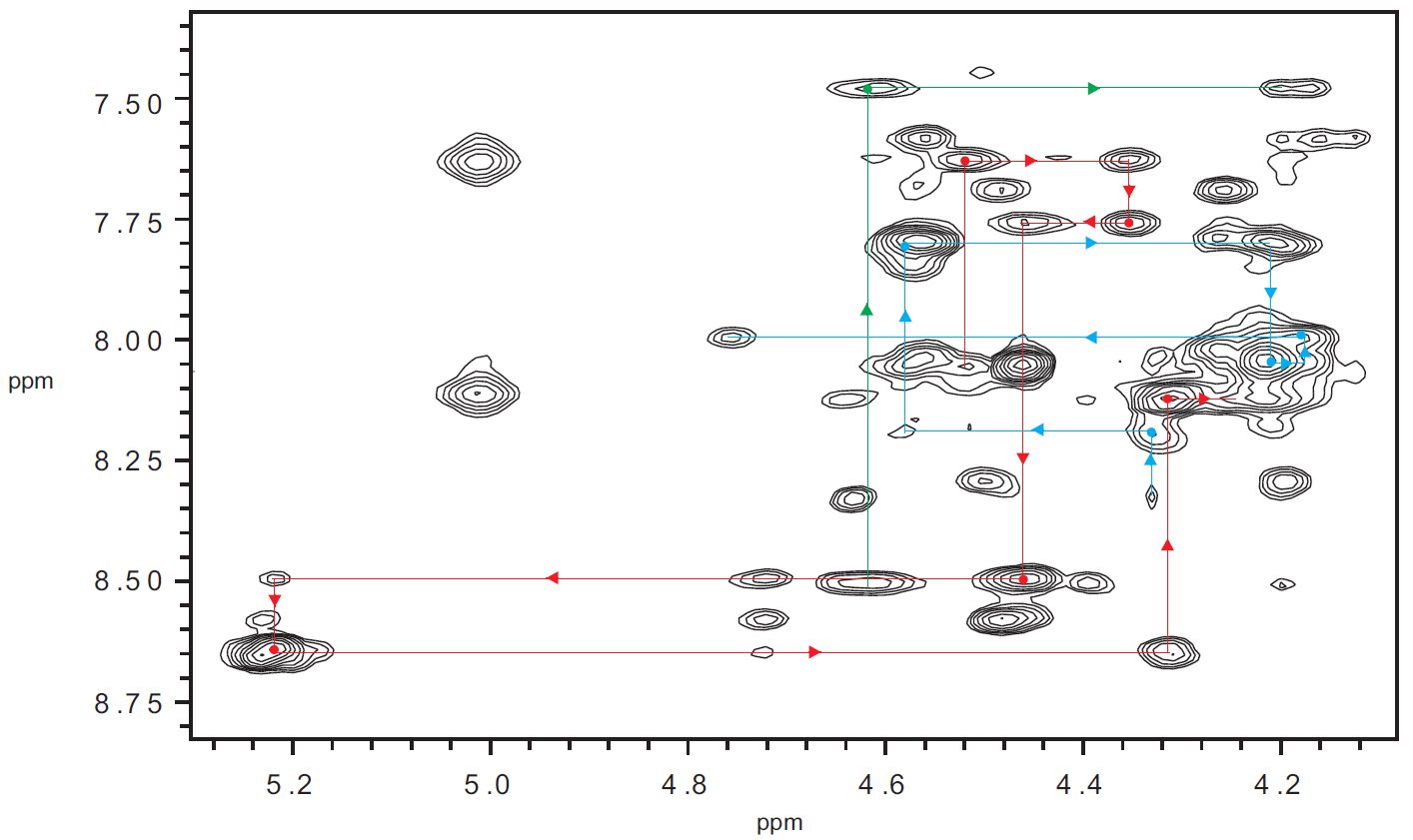
Jak zostało wspomniane we wstępie, nowoczesna spektroskopia magnetycznego rezonansu jądrowego daje potężne narzędzia do badania strukturprzestrzennych złożonych związków chemicznych. Są nimi wielowymiarowe eksperymenty, które dostarczają niezbędnych informacji strukturalnych. Poniżej wymienione zostaną tylko niektóre z technik. Należy mieć świadomość, że aby ustalić strukturę większego biopolimeru należy zarejestrować serię widm trójwymiarowych. Ze względu na fakt, iż tematyka jest bardzo obszerna, w niniejszym artykule przedstawione zastaną tylko techniki dwuwymiarowe, które są wystarczające do wyznaczenia struktury większości badanych peptydów. Podstawowym i analizowanym w pierwszej kolejności eksperymentem jest widmo TOCSY (ang. Totally Correlated Spectroscopy). Technika ta pozwala na obserwację tzw. pojedynczych układów spinowych –rys. 5, z których zbudowana jest cząsteczka. Głównie na podstawie tego widma przypisuje się przesunięcia chemiczne poszczególnym protonom – rys. 6.
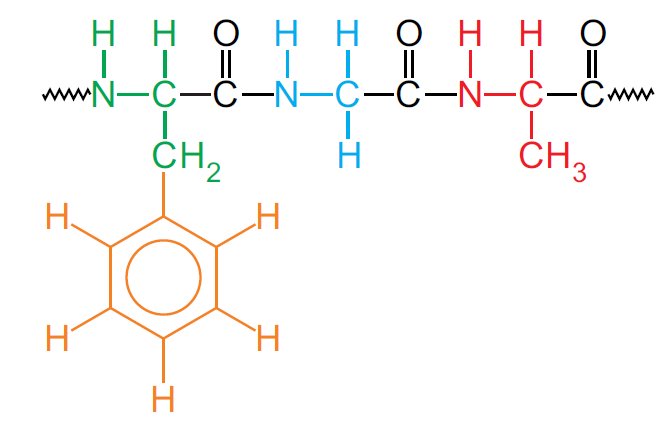
Rysunek 5. Przedstawienie idei układów spinowych. Phe (zielony i pomarańczowy) – dwa układy spinowe, Gly (niebieski) – jeden układ spinowy, Ala (czerwony) – jeden układ spinowy, C=O (czarny) – grupa rozdzielająca poszczególne układy spinowe.
Rysunek 6. Fragment diagnostyczny widma TOCSY zarejestrowanego dla 18 aminokwasowego peptydu. Czerwonymi liniami zaznaczono widoczne układy spinowe.
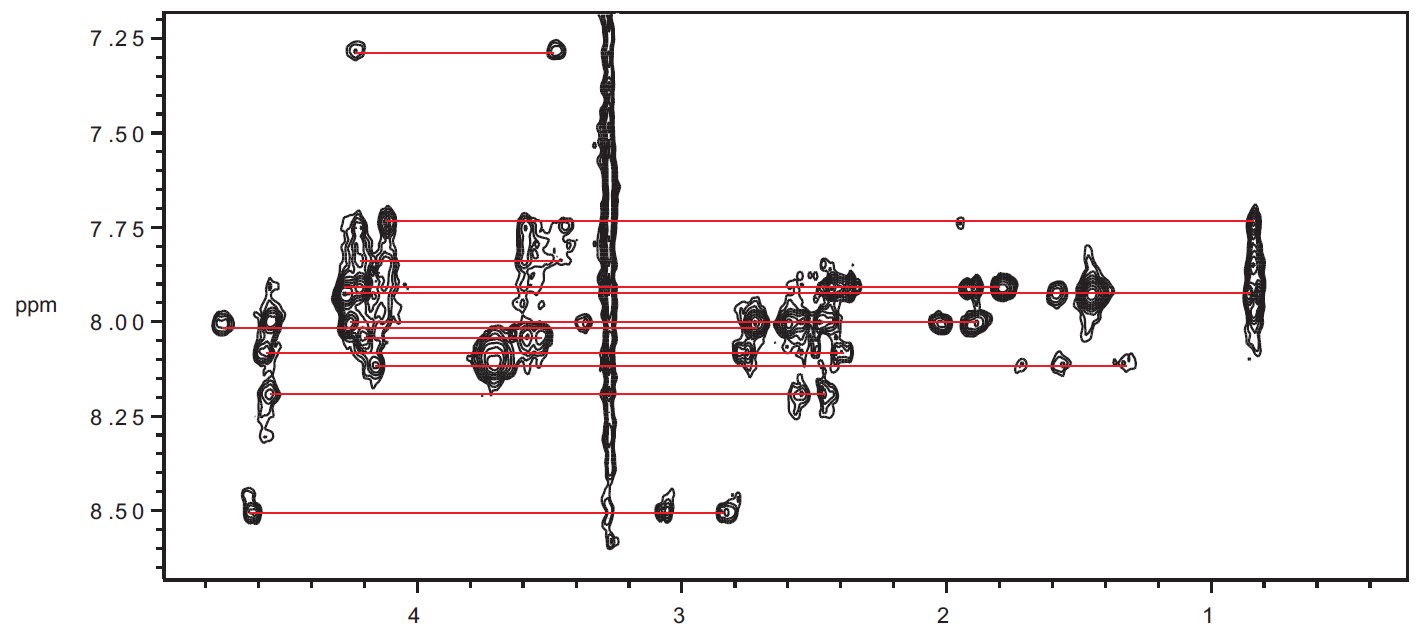
Kolejną techniką jest NOESY (ang. Nuclear Overhauser Effect Spectroscopy). Na widmie tym widoczne są sprzężenia pomiędzy protonami znajdującymi się w odległości około 5Å od siebie. To z tego widma pochodzą wszelkie niezbędne informacje dotyczące struktury badanej cząsteczki. Informacje te to: potwierdzenie sekwencji aminokwasowej na podstawie sprzężeń sekwencyjnych – rys. 7, ustalenie geometrii wiązania peptydowego, zebranie tzw. efektów NOE mówiących o odległościach międzyprotonowych (opis w dalszej części artykułu).
Rysunek 7. Region diagnostyczny widma NOESY zarejestrowanego dla 18 aminokwasowego peptydu. Na widmie widoczne są trzy fragmenty łańcuchów peptydowych, w których aminokwasy połączone są ze sobą wiązaniami peptydowymi zawierającymi protony HN i Hα.
Poznanie konformacji łańcuchagłównego polipeptydu wiąże się ze znajomością kątów wiązania peptydowego. O ile geometrię cis lub trans ustalić można na podstawie widma NOESY to o wartości kąta ϕ mówi wicynalna stała sprzężenia pomiędzy protonami HN i Hα. DQF-COSY to technika pozwalająca na wyznaczenie tychstałych. Oprócz opisanych widm homojądrowych wykorzystywane są również eksperymenty heterokorelacyjne HSQC (ang. Heteronuclear Single Quantum Correlation Spectroscopy) oraz HMBC (ang. Heteronuclear Multiple Bond Coherence). Pierwszy z nich pokazuje sprzężenia pomiędzyheteroatomami C lub N połączonymi z konkretnym protonem, drugi z kolei sprzężenia pomiędzy protonem a węglami oddalonymi o dwa lub trzy wiązania chemiczne.
Peptydy i białka w spektroskopii MRJ
Wysokorozdzielcze pomiary magnetycznego rezonansu jądrowego pozwalają zaobserwować subtelną strukturę multipletów sygnałów rezonansowych. Częstotliwość podstawowa spektrometru jest zatem warunkiem determinującym jakość widm. W przypadku peptydów i białek, gdzie na widmach obecnych jest wiele sygnałów, wysokie pole magnetyczne jest koniecznością. Na rysunku 8 pokazane są zakresy przesunięć chemicznych protonów znajdujących się w cząsteczkach peptydów i białek.
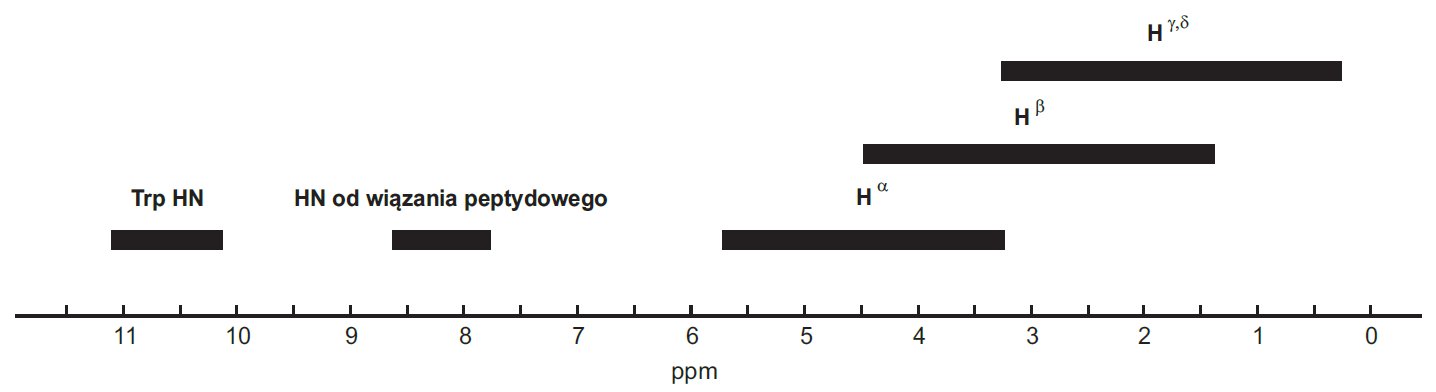
Rysunek 8. Zakresy przesunięć chemicznych niektórych protonów występujących w peptydach i białkach.
Multipletowość jest efektem sprzężenia typu spin-spin pomiędzy sąsiadującymi jądrami atomowymi. W przypadku peptydów i białek jednym z czynników określających kształt łańcucha głównego jest wartość kąta ϕ – rys. 9. Uzyskuje się ją rozwiązując równanie Bystrova-Karpulusa na podstawie wyznaczonej stałej sprzężenia pomiędzy protonami HN i Hα– rys. 10.

Rysunek 9. Kąty torsyjne definiujące geometrię wiązania peptydowego.
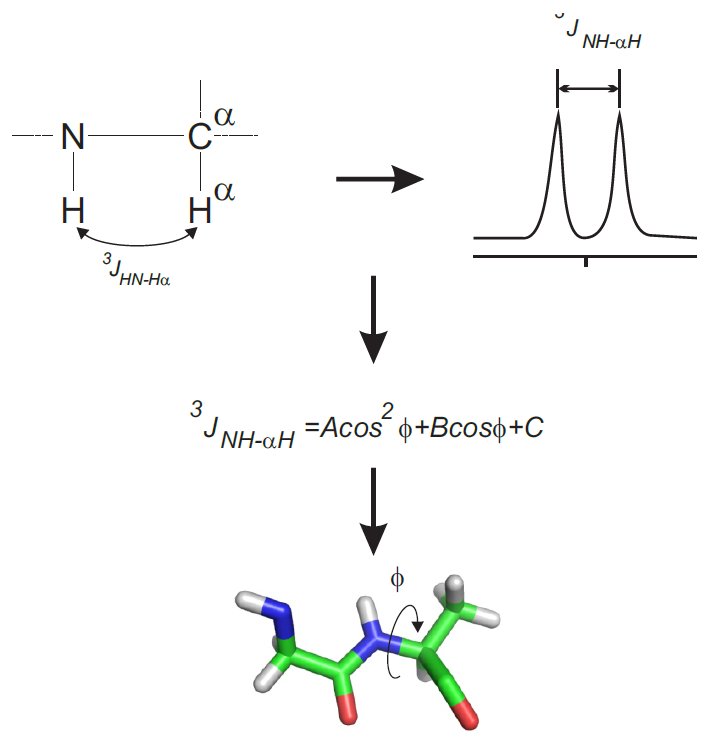
Rysunek 10. Algorytm wyznaczania kąta ϕ wiązania peptydowego.
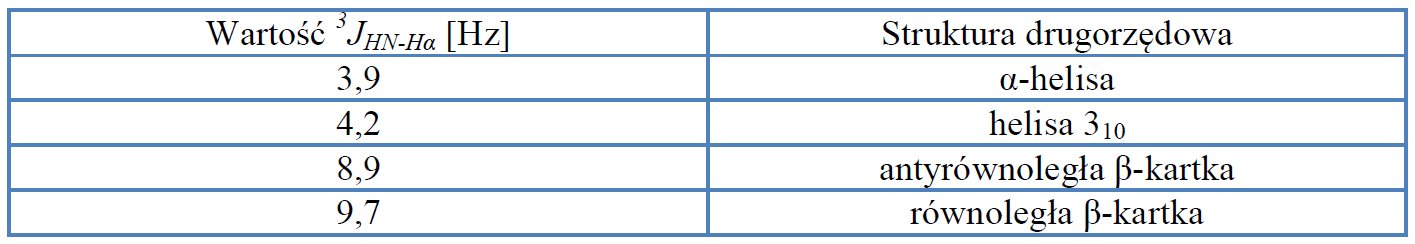
Wspomnieć należy, że wartości stałej sprzężenia 3JHN-Hα odpowiadają konkretnym elementom struktury drugorzędowej peptydów lub białek – tabela 1. Można je odczytać z protonowego widma jednowymiarowego lub z widma DQF-COSY.
Tabela 1. Wartości wicynalnych stałych sprzężenia i odpowiadające im struktury drugorzędowe peptydów i białek.
Wiele informacji strukturalnych uzyskać można z dwuwymiarowego widma NOESY. Znajomość intensywności sygnałów (tzw. efektów NOE - jądrowy efekt Overhausera, ang. Nuclear Overhauser Effect) pochodzących od par spinów, przeliczyć można na odległości międzyprotonowe. Te z kolei są następnym czynnikiem determinującym strukturę przestrzenną biopolimeru. Na rysunku 11 przedstawione są wzory krótkozasięgowych efektów NOE i odpowiadające im elementy struktur drugorzędowych. Występowanie sprzężeń pomiędzy protonami Hα dwóch sąsiadujących ze sobą aminokwasów świadczy o geometrii cis wiązania peptydowego.
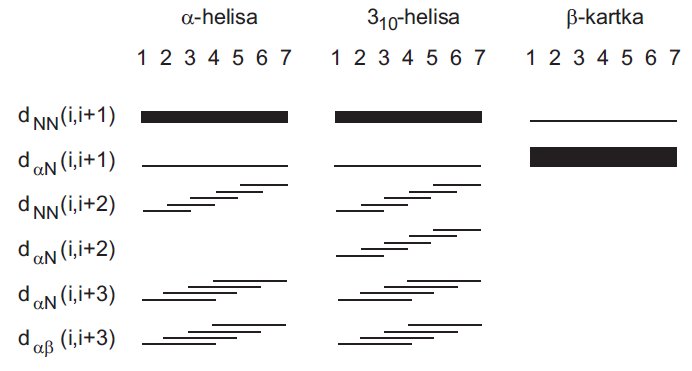
Rysunek 11. Wzór efektów NOE charakterystycznych dla określonych struktur drugorzędowych.
Opisując budowę peptydu lub białka, ważne jest również położenie wiązań wodorowych utworzonych przez atomy łańcucha głównego. Wiązania te stabilizują strukturę przestrzenną całej cząsteczki oraz są one odpowiedzialne za utrzymanie struktur drugorzędowych. Położenie i siłę wiązań wodorowych oznacza się wyznaczając współczynniki temperaturowe dla protonów HN, które są donorami tych wiązań. Technicznie wykonuje się serię widm jednowymiarowych w różnych temperaturach, a następnie oznacza różnice w przesunięciach chemicznych względem temperatury – rys. 12.
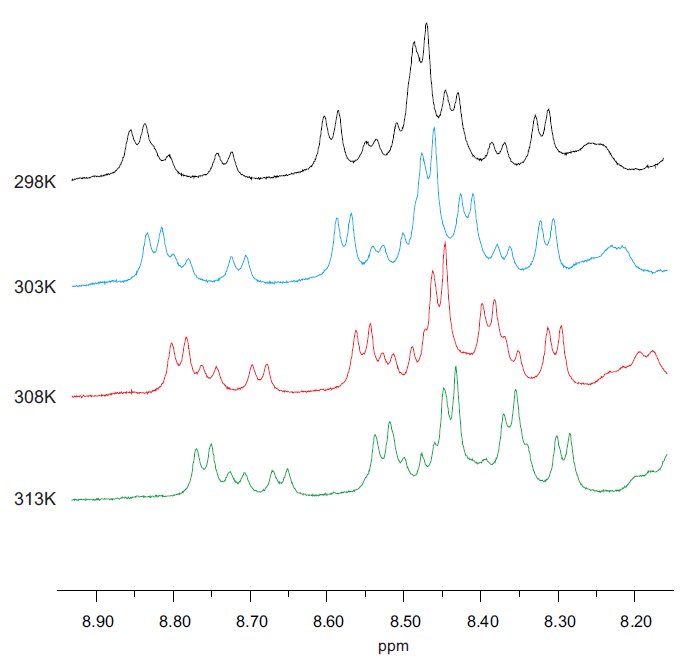
Rysunek 12. Seria widm temperaturowych pokazujących zmianę położenia sygnałów protonów HN pod wpływem temperatury.
Algorytm oznaczania struktury przestrzennej peptydu lub białka
Wszystkie dane uzyskane z widm magnetycznego rezonansu jądrowego stanowią więzy, które wprowadza się do komputerowego programu obliczeniowego. Oczywiście wymagana jest również znajomość struktury pierwszorzędowej czyli sekwencja aminokwasowa, położenie mostków disulfidowych oraz wszelkiego rodzaju modyfikacje (cyklizacja, podstawienie w łańcuchu bocznym, itp). Obliczenia dynamiki molekularnej prowadzące do wygenerowania trójwymiarowego modelu cząsteczki, mogą być przeprowadzane w przestrzeni kartezjańskiej, gdzie każdy atom traktowany jest indywidualnie, a struktura jest „dyktowana” przez potencjały pola siłowego lub z wykorzystaniem sztywnej geometrii walencyjnej gdzie strukturaposzczególnych reszt aminokwasowych jest z góry zdefiniowana, a w celu wygenerowania struktury badanego obiektu zaburzane są jedynie kąty walencyjne. Rysunek 13 pokazuje schemat klasycznego podejścia stosowanego podczas oznaczania struktur przestrzennych peptydów i małych białek.
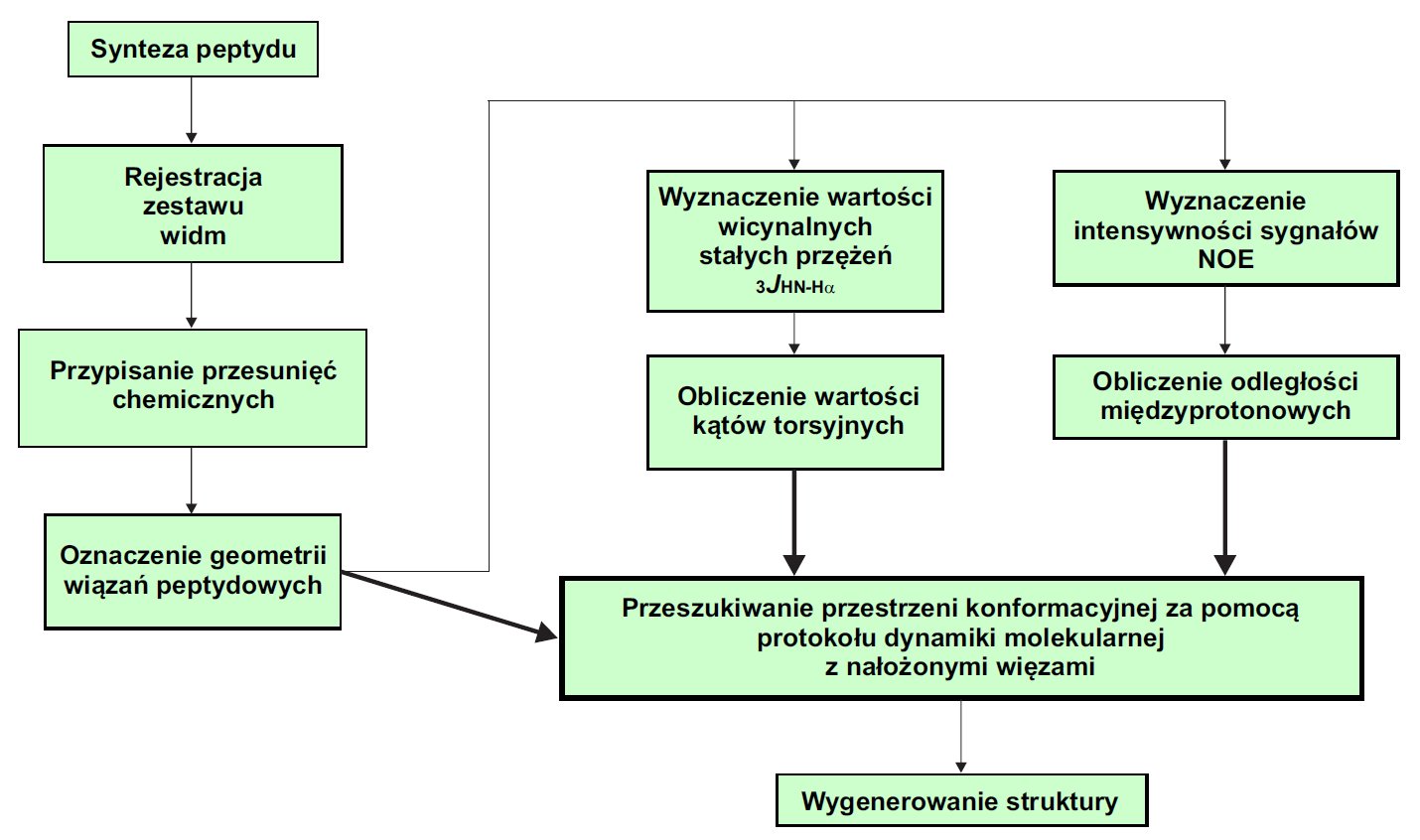
Rysunek 13. Algorytm pokazujący klasyczne podejście do wyznaczania struktury przestrzennej peptydu.
Polecana literatura:
1. K. Brzozowski, Mass Spectrometry and Chosen Spectroscopic Methods of Conformational Studies of Peptides and Proteins, http://lismidos.strony.ug.edu.pl/pliki/skrypty/Brzozowski_Mass_spectrometry.pdf
2. J.N.S. Evans. Biomolecular NMR Spectroscopy, Oxford University Press Inc., New York 1996.
3. G.A. Morris, J.W. Emsley. Multidimensional NMR Methods for the Solution State, John Wiley & Sons Ltd. 2010.
4. J. Keeler. Understanding NMR Spectroscopy, Second Edition, John Wiley & Sons Ltd. 2010.


