


Dawid Jagieła, Sylwia Łuczak
Zakład Modelowanie Molekularnego, Wydział Chemii, Uniwersytet Gdański
strony wersji drukowanej: 27-30
strony wersji drukowanej: 27-30
Chemia od początków swego istnienia operuje modelami, którymi tłumaczymy zachodzące procesy chemiczne. Chcąc zobrazować przebieg reakcji chemicznej, czyli zrywanie lub tworzenie się wiązań chemik mówi o modelu atomowym zbudowanym z dodatnio naładowanego jądra atomowego, oraz krążących wokół niego ujemnie naładowanych elektronów. Organizacja tych elementów na poziomie kwantowym w układy o niższej energii jest właśnie siłą napędową reakcji chemicznej. Jednak nie zawsze chemicy traktują cząsteczki na takim poziomie złożoności. Najczęściej wystarczy posłużyć się modelem cząsteczki, w której atomy traktowane są, jako sztywne kule, a wiązania chemiczne, jako mniej lub bardziej rozciągliwe sprężyny łączące je. Za pomocą takiego podejścia można tłumaczyć wiele zjawisk np. zmiany konformacji cząsteczek oraz dlaczego pewne konformacje bywają bardziej uprzywilejowane od innych. Badając złożone układy tj. kwasy nukleinowe, czy też proteiny stosuje się jeszcze inne modele, w których najmniejszą częścią układu jest pojedynczy nukleotyd lub aminokwas. Jak widzimy w zależności od poziomu, na którym rozpatrujemy problem badawczy można zastosować inny model, który przy obecnym stopniu rozwoju technik komputerowych przekłada się to na zastosowanie odpowiedniego oprogramowania. Na szczęście w obecnych czasach dostęp do mocy obliczeniowej nie stanowi większego problemu, tak, więc możliwości, jakie daje to podejście są olbrzymie.
Projektowanie związków aktywnych biologicznie
Niewątpliwie jednym z motorów napędowych współczesnej chemii jest przemysł farmaceutyczny. Mimo sporych nakładów finansowych, jakimi dysponują koncerny farmaceutyczne nadal głównym kryterium decydującym o sukcesie jest czas, gdyż wprowadzenie na rynek nowego leku trwa średnio 30 lat. Stosując komputerowe wspomaganie projektowania leków (computer aided drug design) można obniżyć koszty oraz znacząco przyspieszyć proces projektowania farmaceutyku. Istnieją dwie podstawowe gałęzie projektowania leków w zależności od tego, jakie informacje posiadamy. Pierwsza to projektowanie oparte o strukturę znanych ligandów (ligand based design), którą stosuje się, wtedy kiedy nie posiadamy informacji na temat miejsca wiążącego receptora, a jedynie znamy budowę innych ligandów z nim oddziałujących. Druga metoda to projektowanie oparte o strukturę receptora (receptor based design), którą stosuje się, wtedy kiedy posiadamy dane na temat budowy receptora, oddziaływań występujących w miejscu wiążącym oraz zmianach konformacyjnych, jakim on ulega (często zmiana konformacji receptora np. poprzez fosforylację prowadzi dopiero do jego aktywacji). Projektowanie oparte o strukturę znanych ligandów sprowadza się do zbudowania farmakoforu, czyli modelu zawierającego przestrzenne relacje pomiędzy grupami wspólnymi dla znanych ligandów (Rys. 1).
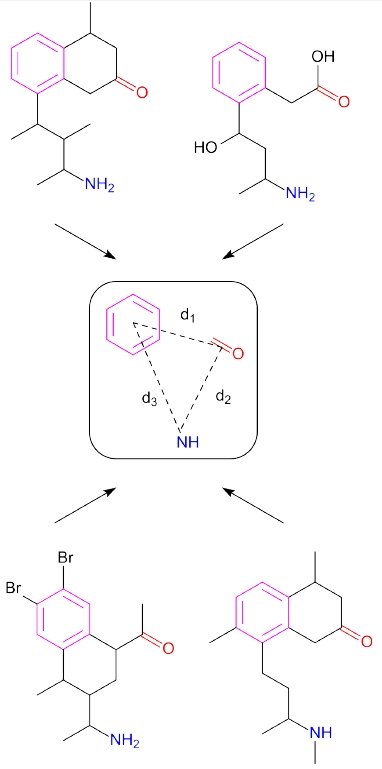
Rysunek 1: Farmakofor zbudowany na podstawie czterech ligandów zawierających pierścień aromatyczny(miejsce oddziaływań hydrofobowych), grupę aminową (donor protonu) oraz karbonyl (akceptor protonu).
Posiadając bazę danych ligandów możliwe staje się wyszukanie potencjalnych kandydatów na lek dzięki porównaniu związków z bazy pod kątem zgodności z farmakoforem. Sam farmakofor można również wy-korzystać do wygenerowania ligandów de novo w oparciu o bazę fragmentów molekularnych. Niezależnie od postępowania otrzymuje się potencjalnych kandydatów na związki wiodące (lead compounds), które następnie przechodzą etap wnikliwej analizy oraz szereg modyfikacji tj. zmiana podstawników mających na celu optymalizację działania farmaceutyku.
Kieszeń wiążąca, a projektowanie związków
Dobrym podejściem na poziomie modyfikowania związku wiodącego jest uzupełnienie wiedzy o dodatkowe informacje na temat budowy miejsca wiążącego (Rys. 2).
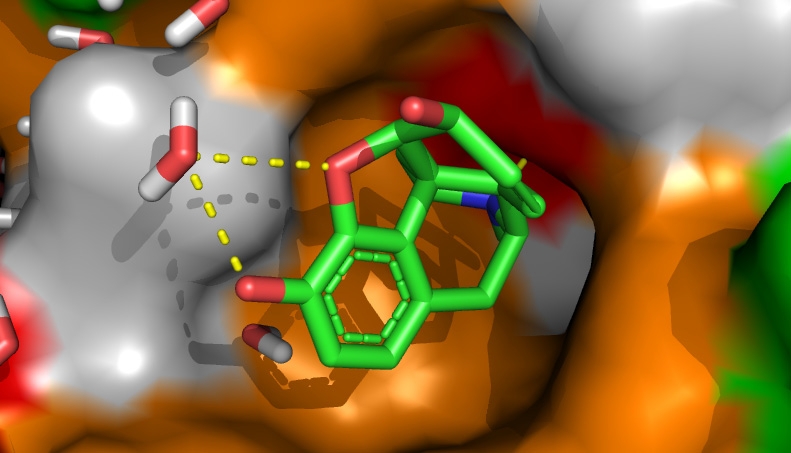
Rysunek 2: Przykładowe miejsce wiążące ligand(morfinę). Kolorem oznaczono odpowiednio reszt hydrofobowych (szare), aromatycznych (pomarańczowe), polarnych(zielone) oraz ujemnie naładowanych (czerwony).
Kiedy struktura receptora nie jest znana można posiłkować się modelowaniem homologicznym. To postępowanie bazuje na podobieństwie w budowie białek u organizmów, które są ze sobą spokrewnione. Informacja na temat miejsca wiążącego w receptorze jest istotnym czynnikiem wpływającym na proces modyfikowania związku wiodącego. Znajomość występujących oddziaływań oraz rozmiarów kieszeni wiążącej pozwala na racjonalne modyfikowanie związków (nie ma przecież sensu wprowadzanie grup posiadających zawadę steryczną, dzięki której związek nigdy nie zwiążę się z receptorem docelowym!). W celu oceny położenia oraz siły oddziaływań liganda z receptorem stosuje się metodę dokowania (komputerowe umieszczenie liganda w miejscu wiążącym). Z reguły dokowaniu poddaje się od kilku do nawet kilkunastu tysięcy ligandów dla każdego wykonując je wielokrotne. Każdy z powstałych kompleksów ocenia się pod kątem założonych wcześniej czynników (energia oddziaływań, czy brak zawad sterycznych). Dzięki wspomnianym powyżej metodom komputerowym znacząco zawęża się grupę potencjalnych leków, które po zsyntezowaniu z dużym prawdopodobieństwem będą posiadały oczekiwane właściwości farmakologiczne.
Podejrzeć cząsteczkę
Jeżeli chcielibyśmy uchwycić jakiś piękny widok, bądź podniosłe wydarzenie sięgniemy po aparat fotograficzny lub kamerę video. Czy w przypadku biomolekuły jest to możliwe? Mimo iż bez problemu dostaniemy wysokiej klasy aparat nawet on nie będzie w stanie sfotografować momentu, w którym cząsteczka morfiny przyłącza się do receptora. Z pomocą przychodzą tu wyrafinowane metody pomiarowe tj. rentgenografia strukturalna, która jest odpowiednikiem aparatu fotograficznego w świecie cząsteczek. Niestety nawet i najlepszy dyfraktometr jest w stanie tylko uchwycić chwilę z życia molekuły, ale nie pokaże jej dynamicznego charakteru. Przypomina to próbę fotografowania muchy w zamkniętym pomieszczeniu – na zdjęciu widać ją zamrożoną w powietrzu, a nie widać samej czynności latania. Robienie serii zdjęć, sekunda po sekundzie, pozwala nam wnioskować tylko, że mucha częściej przebywała w okolicach lampy na suficie, niż podłogi, ale dalej nie wiemy jak mucha poruszała się po pokoju. Molekularnym odpowiednikiem serii stopklatek są techniki magnetycznego rezonansu jądrowego (Nuclear Magnetic Resonance). W wyniku skomplikowanych pomiarów NMR oraz cyfrowej obróbki danych otrzymujemy zbiór położeń cząsteczki, ale ciągle nie wiemy, co się z nią działo między jednym stanem, a drugim. Pełen obraz możemy otrzymać łącząc wiedzę o poszczególnych stanach cząsteczki z prawdopodobnym przejściem (ruchem), jaki mógł zajść pomiędzy stanami – tu nieodzownym narzędziem jest dynamika molekularna. Sama metoda polega na przeprowadzeniu symulacji układu molekuł, w której iteracyjnie w danym kroku czasowym obliczane są siły działające na poszczególne atomy wprawiające je w ruch. Z punktu widzenia chemii komputerowej numeryczne rozwiązywanie takiej symulacji jest bardzo szybkie, gdyż w elementarnej wersji sprowadza się do całkowania równań ruchu Newtona.
Tania dynamika molekularna
Jakie jest praktyczne zastosowanie dynamiki molekularnej? Otóż można wymodelować np. zachowanie się wszelkiego rodzaju związków amfifilowych w wodzie (Rys. 3).
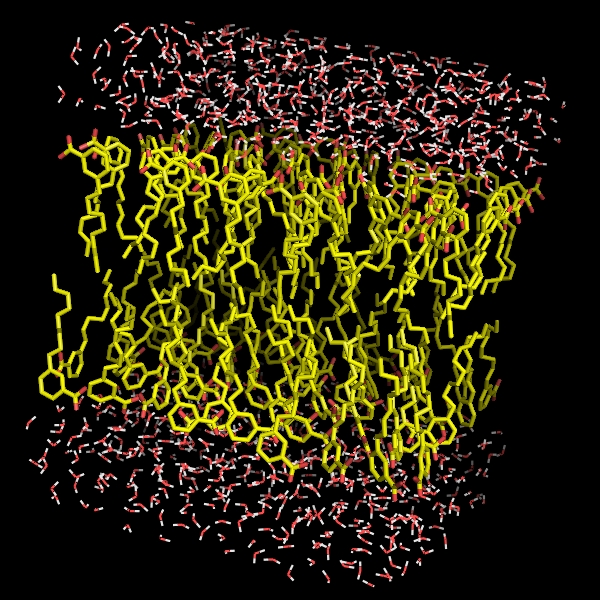
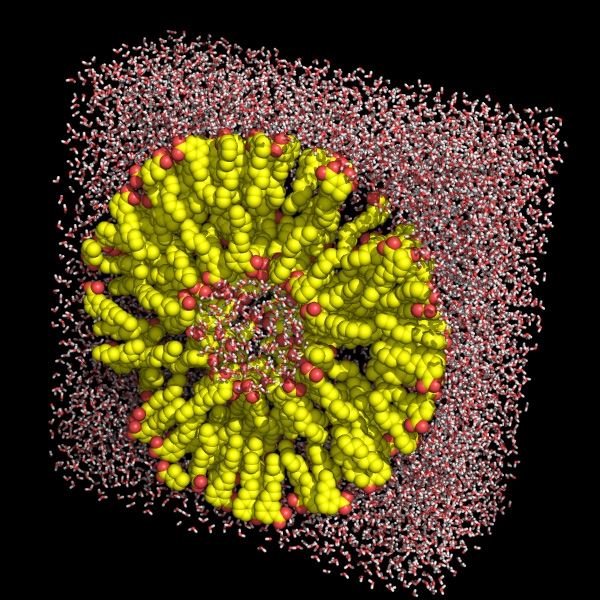
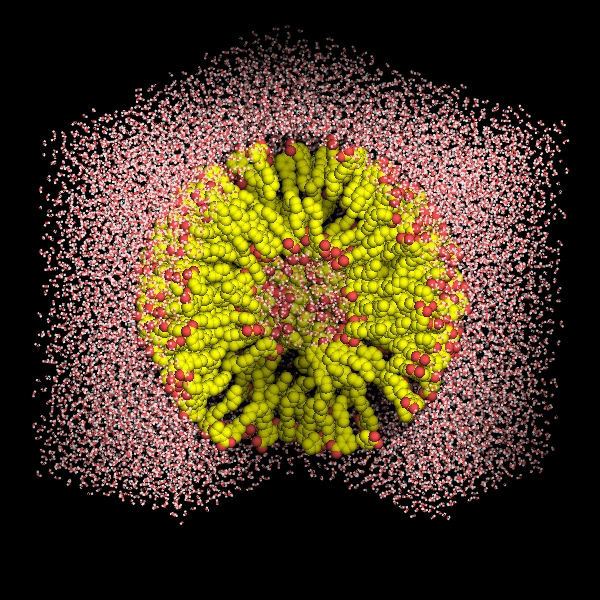
Rysunek 3: Przykładowe modele układów amfifilowych w wodzie.
Rynek produktów higieny osobistej szacowany jest na ok. 40 miliardów dolarów, z czego rynek szamponów to ok 10 miliardów dolarów. Znajomość procesów zachodzących na poziomie molekularnym pozwala na projektowanie lepszych surfaktantów, czyli związków powierzchniowo czynnych. Symulacje tego typu są o tyle ciekawe, że w ciągu ostatnich kilku lat modelarze otrzymali do rąk potężne narzędzie – możliwość prowadzenia obliczeń na kartach graficznych. Porównując dynamikę molekularną prowadzoną na klasycznie zwykłym procesorze, która trwa kilka dni z tą samą symulacją prowadzoną na karcie graficznej, która trwa godzinę uświadamiamy sobie, jaka moc obliczeniowa znajduje się w rękach posiadacza komputera multimedialnego. Kolejnym przykładem zastosowania dynamiki molekularnej jest projektowanie antybiotyków. Działanie części z nich opiera się na zakłócaniu funkcjonowania struktur komórkowych np. błony komórkowej bakterii, tak więc aby dany związek działał powinien posiadać pewne powinowactwo do błony. Możliwe jest zbudowanie modelu błony komórkowej, a następnie przeprowadzenie dynamiki molekularnej dla potencjalnego antybiotyku, by sprawdzić czy aby, a jeśli tak to w jaki sposób następuje penetracja błony komórkowej. Takie podejście jest wielokrotnie tańsze niż żmudna synteza i oczy-szczanie, a następnie testy biologiczne. Przykłady zastosowania modelowania molekularnego można wymieniać w nieskończoność, a i tak nie pokazałoby to pełni użyteczności tej techniki. Jest to szybkie, oszczędne i czyste narzędzie w pracy naukowej nie tylko chemików.


