


Tomasz Bieńkowski
Laboratorium Masdiag
strony wersji drukowanej: 50-55

Pod koniec XX wieku rozpoczął się projekt poznania ludzkiego genomu (Human Genom Project). Zakończył się on w 2003 roku. Środowisko naukowe wiązało z nim wielkie nadzieje, związane szczególnie z diagnozowaniem chorób. Niestety, nie do końca się one spełniły. W wielu przypadkach na podstawie badań genetycznych można zdiagnozować wybrane choroby, ale w jeszcze większej ilości przypadków okazało się, że genom wskazuje na predyspozycje danego organizmu, natomiast o faktycznym stanie organizmu więcej może powiedzieć metabolomika, proteomika, lipidomika i glikomika czyli nauki, które zajmują się przemianami i stanem metabolitów oraz różnych grup biocząsteczek. W dziedzinach tych spektrometria mas ma wiele zastosowań. W poniższym artykule zostaną zaprezentowane wybrane zagadnienia dotyczące zastosowania spektrometrii mas w proteomice.
strony wersji drukowanej: 50-55
Pod koniec XX wieku rozpoczął się projekt poznania ludzkiego genomu (Human Genom Project). Zakończył się on w 2003 roku. Środowisko naukowe wiązało z nim wielkie nadzieje, związane szczególnie z diagnozowaniem chorób. Niestety, nie do końca się one spełniły. W wielu przypadkach na podstawie badań genetycznych można zdiagnozować wybrane choroby, ale w jeszcze większej ilości przypadków okazało się, że genom wskazuje na predyspozycje danego organizmu, natomiast o faktycznym stanie organizmu więcej może powiedzieć metabolomika, proteomika, lipidomika i glikomika czyli nauki, które zajmują się przemianami i stanem metabolitów oraz różnych grup biocząsteczek. W dziedzinach tych spektrometria mas ma wiele zastosowań. W poniższym artykule zostaną zaprezentowane wybrane zagadnienia dotyczące zastosowania spektrometrii mas w proteomice.
 Pierwszym zastosowaniem spektrometrii mas w proteomice była identyfikacja białek. Istnieją dwie strategie identyfikacji, z góry na dół (top down) czyli analiza sekwencji dla całych białek oraz z dołu do góry (buttom-up), w której badane białko trawi się enzymatycznie i analizę przeprowadza się na poziomie peptydów. Na podstawie oznaczonych peptydów rozpoznaje się białko. Peptydy identyfikuje się głównie na podstawie widm fragmentacyjnych przy użyciu bibliotek składających się z sekwencji białek. Prowadzi się również próby analizy z góry na dół na różnorodnych typach spektrometrów, ale jak na razie strategia ta nie zdobyła zbyt dużej popularności. Zdecydowanie więcej pomiarów prowadzi się stosując metodę z dołu do góry, zarówno przy identyfikacji czystych białek (np. plamek z żeli dwuwymiarowych), jak i skomplikowanych mieszanin. W przypadku czystych białek lub mieszanin kilku białek identyfikację można prowadzić za pomocą MALDI TOF/TOF lub za pomocą tandemowych systemów LC/MS/MS. Natomiast bardziej złożone mieszaniny białek można identyfikować za pomocą wysokorozdzielczych systemów LC/MS/MS typu QqTOF lub Orbitrap. W zależności od tego jak bardzo skomplikowana jest mieszanina pierwszym krokiem jest trawienie enzymatyczne. Następnie wykonuje się bezpośrednią analizę LC/MS/MS lub stosuje się wstępne frakcjonowanie próbki. W czasie analizy wykonywane jest widmo MS wysokiej rozdzielczości, a następnie, dla zdefiniowanej ilości najwyższych sygnałów pochodzących od peptydów (od 10 do 50 w zależności od typu mieszaniny i rodzaju spektrometru mas), wykonywane są widma fragmentacyjne. Podczas analizy trwającej do kilku godzin możliwa jest identyfikacja kilku tysięcy białek. Metoda ta ma bardzo duży potencjał, ale do uzyskania jak najlepszych wyników potrzebny jest czuły spektrometr charakteryzujący się dużą szybkością zbierania danych w trybie MS/MS. Największym ograniczeniem tej metody jest jej niska powtarzalność, szczególnie dla białek o małym stężeniu. Kluczowym etapem jest wybór jonów, dla których zostanie wykonane widmo fragmentacyjne. W zależności od pomiaru różne jony dają najwyższe wygnały w widmie MS i dla różnych peptydów wykonywane są widma fragmentacyjne. Szacuje się, że powtarzalność tego typu pomiarów wynosi około 60%.
Pierwszym zastosowaniem spektrometrii mas w proteomice była identyfikacja białek. Istnieją dwie strategie identyfikacji, z góry na dół (top down) czyli analiza sekwencji dla całych białek oraz z dołu do góry (buttom-up), w której badane białko trawi się enzymatycznie i analizę przeprowadza się na poziomie peptydów. Na podstawie oznaczonych peptydów rozpoznaje się białko. Peptydy identyfikuje się głównie na podstawie widm fragmentacyjnych przy użyciu bibliotek składających się z sekwencji białek. Prowadzi się również próby analizy z góry na dół na różnorodnych typach spektrometrów, ale jak na razie strategia ta nie zdobyła zbyt dużej popularności. Zdecydowanie więcej pomiarów prowadzi się stosując metodę z dołu do góry, zarówno przy identyfikacji czystych białek (np. plamek z żeli dwuwymiarowych), jak i skomplikowanych mieszanin. W przypadku czystych białek lub mieszanin kilku białek identyfikację można prowadzić za pomocą MALDI TOF/TOF lub za pomocą tandemowych systemów LC/MS/MS. Natomiast bardziej złożone mieszaniny białek można identyfikować za pomocą wysokorozdzielczych systemów LC/MS/MS typu QqTOF lub Orbitrap. W zależności od tego jak bardzo skomplikowana jest mieszanina pierwszym krokiem jest trawienie enzymatyczne. Następnie wykonuje się bezpośrednią analizę LC/MS/MS lub stosuje się wstępne frakcjonowanie próbki. W czasie analizy wykonywane jest widmo MS wysokiej rozdzielczości, a następnie, dla zdefiniowanej ilości najwyższych sygnałów pochodzących od peptydów (od 10 do 50 w zależności od typu mieszaniny i rodzaju spektrometru mas), wykonywane są widma fragmentacyjne. Podczas analizy trwającej do kilku godzin możliwa jest identyfikacja kilku tysięcy białek. Metoda ta ma bardzo duży potencjał, ale do uzyskania jak najlepszych wyników potrzebny jest czuły spektrometr charakteryzujący się dużą szybkością zbierania danych w trybie MS/MS. Największym ograniczeniem tej metody jest jej niska powtarzalność, szczególnie dla białek o małym stężeniu. Kluczowym etapem jest wybór jonów, dla których zostanie wykonane widmo fragmentacyjne. W zależności od pomiaru różne jony dają najwyższe wygnały w widmie MS i dla różnych peptydów wykonywane są widma fragmentacyjne. Szacuje się, że powtarzalność tego typu pomiarów wynosi około 60%.Identyfikacja białek w dalszym ciągu jest jednym z popularniejszych zastosowań spektrometrii mas, ale coraz częściej wykorzystuje się tą technikę do badania struktury białek i do analizy ilościowej.
Jedną z metod badania trzecio- i czwartorzędowej struktury białka jest mierzenie stopnia wymiany wodoru na deuter HDX (Hydrogen Deuter Exchange) przy pomocy spektrometrii mas. Pomiary wykonuje się umieszczając badane białko w roztworze deuterowanej wody D2O w kontrolowanych warunkach. Następuje wymiana wodoru na deuter, a po zatrzymaniu reakcji wymiany poprzez obniżenie pH do 2,5 i obniżenie temperatury do 0oC wykonuje się pomiar LC/MS/MS na spektrometrze wysokiej rozdzielczości. Widmo można wykonywać dla cząsteczki białka lub po szybkiej hydrolizie pepsyną na poziomie peptydowym. W cząsteczkach białka istnieją 3 grupy atomów wodoru. Pierwsza grupa to atomy związane z atomami węgla. Wiązanie węgiel wodór jest bardzo trwałe i wymiana wodoru na deuter jest zbyt wolna, żeby ją zaobserwować. Kolejna grupa to atomy wodoru występujące w grupach funkcyjnych, związane z atomem azotu lub tlenu (-NH3, -OH, -COOH). Wymiana tych atomów jest z kolei bardzo szybka i w momencie, gdy reakcja jest zatrzymywana w roztworach zawierających H2O, atomy deuteru wymieniane są z powrotem na atomy wodoru. Ostatnią grupę stanowią atomy wodoru związane z amidowym atomem azotu w wiązaniu peptydowym. Szybkość ich wymiany zależy od pH (najwolniejsza jest przy wartość około 2,6) oraz od temperatury. Na szybkość wymiany ma również wpływ położenia grup amidowych. Grupy, które znajdują się na zewnątrz struktury białka i do których cząsteczki rozpuszczalnika mają lepszy „dostęp” ulegają wymianie szybciej, natomiast te, do których cząsteczki rozpuszczalnika nie mają dostępu nie ulegają wymianie. Ponieważ deuter ma masę atomową o 1 większą niż wodór, po wymianie H/D masa cząsteczkowa całego białka wzrasta proporcjonalnie do ilości wymienionych atomów. Porównując wyniki pomiaru przed i po wymianie wodoru na deuter w przypadku analizy całych cząsteczek białka uzyskuje się informację ile atomów wodoru zostało wymienionych, a dla pomiarów na poziomie peptydowym uzyskuje się informacje, w których peptydach i ilu atomów wodoru nastąpiła wymiana, dzięki czemu można stwierdzić, które fragmenty sekwencji białka znajdują się na zewnątrz struktury.
HDX wykorzystuje się do badania różnorodnych właściwości białek. Najważniejsze to:
Badanie interakcji białek z ligandami lub innymi białkami. Pomiar masy cząsteczkowej wykonuje się dla wolnego białka, dla białka związanego z ligandem, z różnymi ligandami lub w różnych warunkach pH. Po przyłączeniu ligandu dostęp dla cząsteczek D2O do miejsc, w których wiąże się ligand zmniejsza się. W zależności od siły wiązania ligandu do białka i ilości miejsc oddziaływania, różna ilość atomów wodoru zostaje wymieniona na deuter. Aby uzyskać dodatkowe informacje na temat miejsca wiązania ligandu, po zatrzymaniu reakcji wymiany, należy poddać cząsteczkę białka hydrolizie pepsyną i pomiary wykonać na poziomie peptydowym. Porównując stopień wymiany H/D przed i po związaniu ligandu można stwierdzić, które fragmenty sekwencji białka odpowiedzialne są za jego wiązanie.
HDX wykorzystuje się również do badania wpływu pH na zmianę konformacji białek. W tym wypadku pomiary są trochę bardziej skomplikowane, ponieważ szybkość wymiany H/D przy wiązaniu amidowym również zależy od pH. Projektując więc doświadczenia, w których zamierza się badać wpływ pH na konformację, należy zmodyfikować czasy prowadzenia wymiany w zależności od pH (Coales SJ, E SY, Lee JE, Ma A, Morrow JA, Hamuro Y. Expansion of time window for mass spectrometric measurement of amide hydrogen/deuterium exchange reactions. Rapid communications in mass spectrometry : RCM. 2010;24(24):3585-92.).
HDX wykorzystuje się ponadto do mapowania epitopów, badania agregacji białek, wpływu mutacji na zmianę konformacji. W wielu wypadkach oprócz relatywnie prostych eksperymentów sprawdzających stopień wymiany H/D przeprowadza się doświadczenia, w których bada się kinetykę procesu wymiany. W porównaniu do techniki NMR zastosowanie spektrometrii mas do mierzenia HDX umożliwia pracę z mniejszą ilością białek, wykonywanie pomiarów dla większych cząsteczek oraz w warunkach zbliżonych do fizjologicznych. Za pomocą spektrometru mas wykonuje się pomiary masy białek i peptydów, jak również widma fragmentacyjne. Wykonując analizy peptydów możliwe jest tylko ustalenie ile atomów wodoru w cząsteczce zostało wymienionych na deuter. Wskazanie, w których wiązaniach peptydowych nastąpiła wymiana nie jest możliwe poprzez wykonanie widma fragmentacyjnego (CID, collision induced dissociation) w komorze zderzeń, ponieważ ponieważ w trakcie tego procesu następuje wymiana atomów wodoru między poszczególnymi wiązaniami.
Aby możliwe było uzyskanie powtarzalnych i znaczących wyników HDX za pomocą spektrometru mas spełniony musi być szereg warunków. Najważniejsze to takie, że zarówno próbki jak i kolumna chromatograficzna muszą mieć możliwość utrzymywania ich w temperaturze 0oC oraz wymagane jest przeprowadzenie szybkiej hydrolizy enzymatycznej pepsyną. W obecnej chwili istnieją komercyjne rozwiązania umożliwiające wykonywanie pomiarów LC/MS/MS. Jeśli chodzi o przygotowanie próbek i sam proces wymiany H/D liderem jest firma Leap Technologies, która dostarcza gotowe do użytku rozwiązania. Do wykonania pomiarów MS i MS/MS potrzebny jest tandemowy spektrometr wysokiej rozdzielczości.
Kolejnym krokiem po identyfikacji białek jest opracowanie metody ilościowego profilowania białek. W przypadku analizy wybranych białek, kiedy analizowanych jest od kilku do kilkudziesięciu peptydów, spektrometry typu potrójny kwadrupol i tryb MRM mogą być wykorzystywane. Tryb MRM (Multiple Reaction Monitoring) jest charakterystyczny dla potrójnego kwadrupola. W pierwszym kwadrupolu w sposób ciągły wybierane są jony o zdefiniowanej wartości m/z. W komorze zderzeń następuje wysokoenergetyczna i wydajna fragmentacja, a drugi analizator kwadrupolowy przepuszcza tylko fragmenty o zdefiniowanej wartości m/z. W ten sposób badany związek rozpoznawany jest na podstawie reakcji fragmentacji. Możliwe jest zbudowanie metody ilościowej zawierającej około 1000 przejść MRM. Aby zwiększyć pewność pomiaru, dla jednego peptydu obserwuje się zazwyczaj kilka przejść MRM, dla jednego białka obserwuje się kilka do kilkunastu peptydów. Daje to możliwość analizowania kilkunastu białek w pojedynczym pomiarze. Wyzwaniem jest wykonanie takiego pomiaru w bardzo skomplikowanej matrycy lub dla dużo większej ilości białek. W przypadku wykonywania pomiarów ilościowych LC/MS/MS białek w skomplikowanej matrycy, np. analiza PSA (antygen sterczowy) w surowicy, pomiar prowadzi się na poziomie peptydowym. Szacuje się, że w surowicy znajduje się około 1 miliona białek i peptydów. Po przeprowadzeniu trawienia trypsyną powstaje niezwykle skomplikowana mieszanina i ryzyko interferencji dla wybranych do pomiarów przejść MRM jest bardzo duże z powodu ilości peptydów w badanej próbce oraz podobnego sposobu fragmentacji peptydów. Dlatego też rozwiązaniem wydaje się stosowanie trybu MRM3, dostępnego na spektrometrach typu QTRAP. Jest to tryb, który polega na dwukrotnej fragmentacji.W pierwszym etapie wybierane są jony macierzyste, a następnie do fragmentacji wybierany jest jeden z powstałych fragmentów. W ten sposób peptyd rozpoznawany jest na podstawie dwukrotnej reakcji fragmentacji, co bardzo zwiększa selektywność.
Kolejnym, wspomnianym wcześniej, wyzwaniem jest profilowanie wielu białek równocześnie. Kiedy chcemy sprawdzić jak zmienia się proteom badanego organizmu pod wpływem danego czynnika, spektrometry typu potrójny kwadrupol nie są wstanie sprostać temu zadaniu. Aby wykonać powtarzalną analizę ilościową w skomplikowanej mieszaninie, wymagane jest wykonanie widma MS/MS dla wszystkich badanych związków. Taką możliwość daje nowa metodyka wykonywania pomiarów – SWATH. Jest to pomiar wykonywany na spektrometrze typu QqTOF, ponieważ niezbędna jest wysoka rozdzielczość i szybkość wy konywania pomiaru. Dodatkowo, czym większa jest czułość pomiaru, tym więcej związków można badać. W dużym uproszczeniu strategia pomiaru polega na podzieleniu zakresu mas prekursorów na przedziały w zakresie 25 m/z. Wszystkie jony macierzystego z tego zakresu są następnie fragmentowane i tworzone jest widmo fragmentacyjne wysokiej rozdzielczości. Widma fragmentacyjne wykonywane są po kolei dla wszystkich przedziałów dla całego zdefiniowanego zakresu mas w sposób cykliczny. W ten sposób zbierane są widma fragmentacyjne dla wszystkich jonizujących się substancji. Dodatkowo pomiędzy cyklami widm fragmentacyjnych wykonywane jest widmo MS wysokiej rozdzielczości (Rysunek 1).
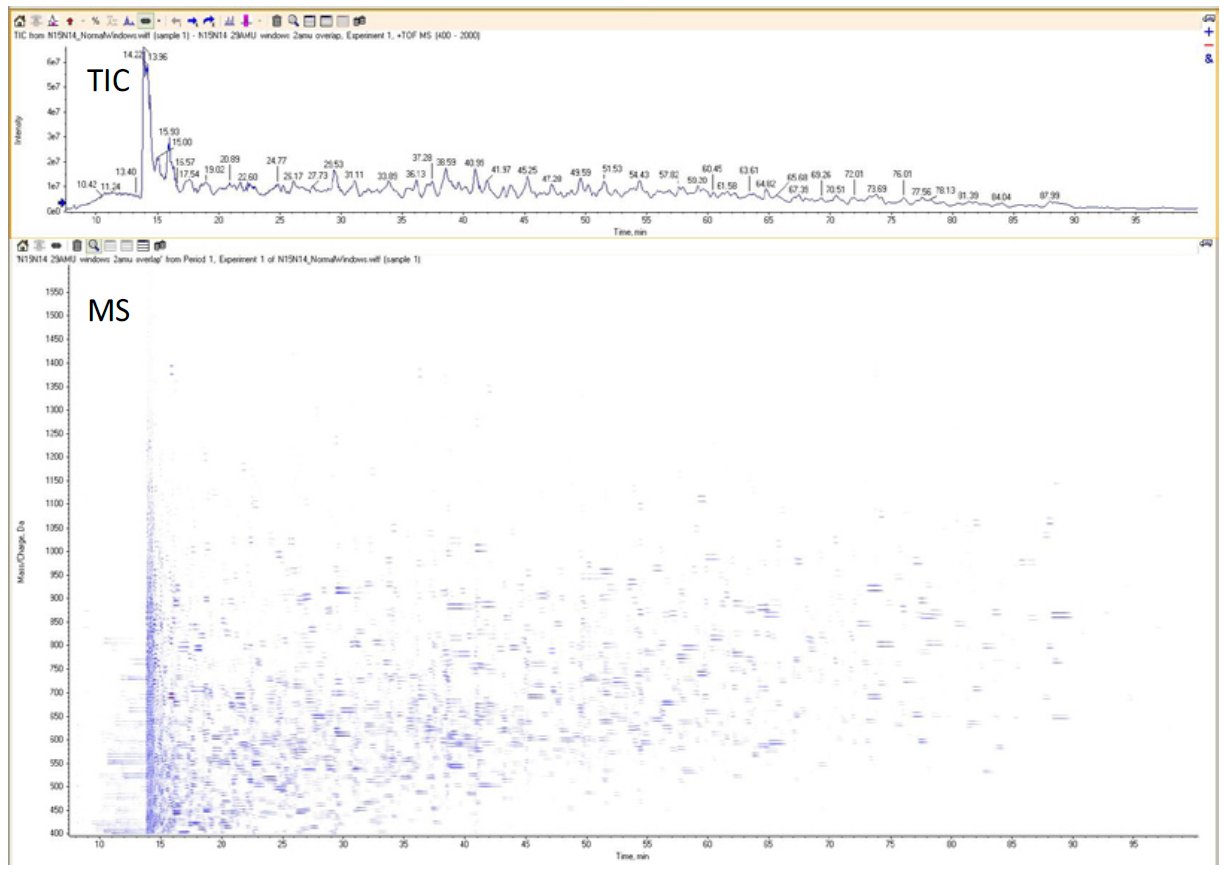
Rysunek 1. Chromatogram całkowitego prądu jonowego i mapa widma MS.
Jako wynik pomiaru całej metody otrzymywany jest bardzo skomplikowany zbiór danych, ponieważ dla każdego zdefiniowanego przedziału wykonywana jest fragmentacja bardzo wielu związków. Jednakże, gdy chcemy sprawdzić czy w próbce znajdował się interesujący nas związek wystarczy wybrać odpowiedni przedział (np. dla peptydu o jonie 2- krotnie naładowanym o m/z 615 będzie to przedział 600 – 625 m/z), w którym powinien znajdować się jon macierzysty i wykonać chromatogram dla specyficznych dla tego związku jonów fragmentacyjnych XIC (Extracted Ion Chromatogram) (Rysunek 2).
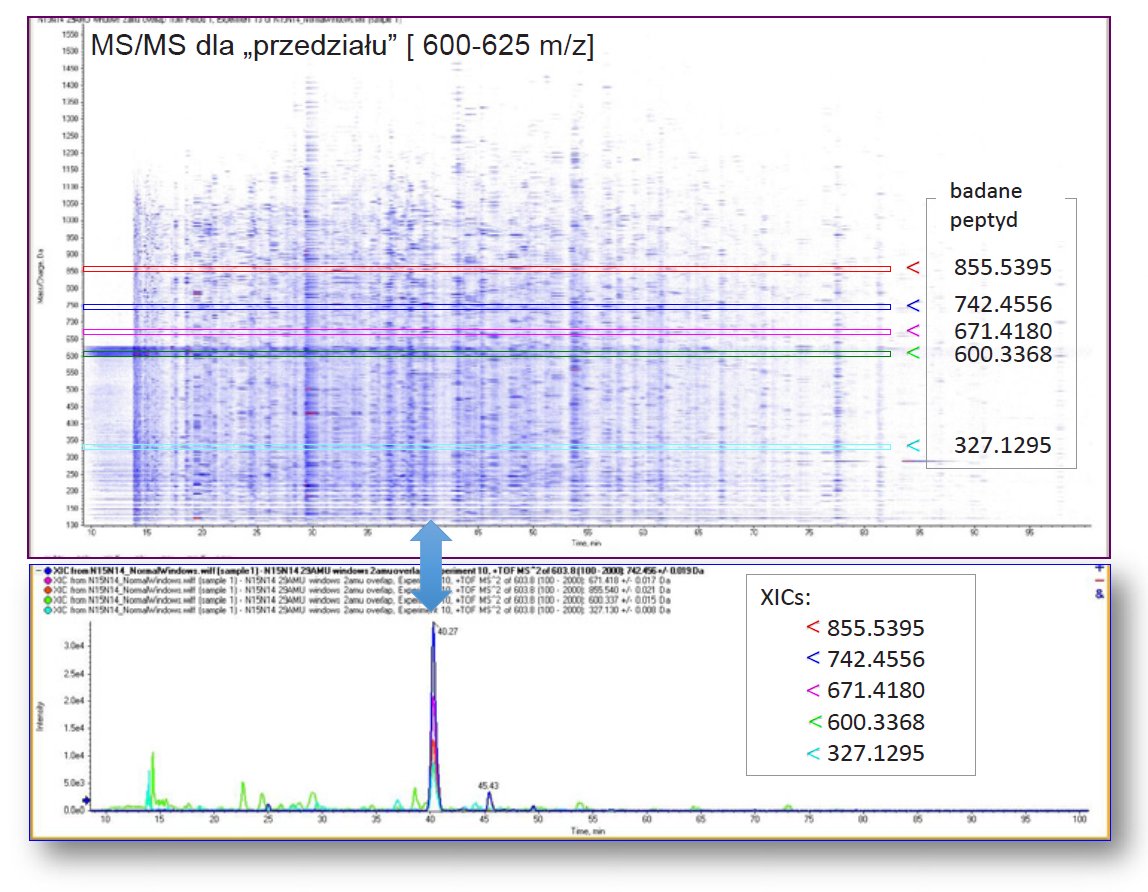
Rysunek 2. Mapa widma MS/MS dla przedziału 600 – 625 m/z oraz chromatogramy dla wybranych jonów fragmentacyjnych.
XIC wykonywane są przy wykorzystaniu dokładnego pomiaru masy z szerokością zakresu 0,01 amu. W wyniku takiej operacji otrzymuje się kilka chromatogramów o wyglądzie zbliżonym do przejść MRM dla wszystkich jonów fragmentacyjnych badanej substancji. Pokrywają się one przy czasie występowania tej substancji. Z symulacji przeprowadzonych dla proteomu drożdży wynika, że selektywność tak zdefiniowanego obserwowania reakcji fragmentacji (25 amu pierwszym analizatorze masy i 0,01 amu w drugim) jest zbliżona do selektywności trybu MRM w potrójnym kwadrupolu (standardowo jest to 0,7 amu dla obu analizatorówmasy). Jednakże przy tak prowadzonym pomiarze nie definiujemy tych przejść przed pomiarem. Wykonywane są one dla wszystkich jonizujących się substancji w próbce. Możliwa jest więc względna analiza ilościowa tysięcy a nawet dziesiątków tysięcy substancji. Warunkiem jest znajomość fragmentacji badanej substancji. Wykonując więc takie pomiary w przypadku próbek proteomicznych, pierwszym etapem jest identyfikacja białek i stworzenie biblioteki peptydów znajdujących się w próbce. Aby zwiększyć ilość zidentyfikowanych związków analizę taką można wykonać wielokrotnie. Dodatkowo w przypadku gdyby dany związek nie został zidentyfikowany, a w wyniku innych doświadczeń lub danych literaturowych okazało się, że dana substancja może się znajdować w analizowanej za pomocą metodyki SWATH próbce, istnieje możliwość sprawdzenia czy rzeczywiście się tam znajdowała. Metodyka SWATH umożliwia więc względną analizę ilościową (profilowanie) bardzo dużej ilości związków oraz tworzy cyfrowy zapis składu próbki i umożliwia retrospektywną analizy danych.
Jeśli zainteresował Państwa temat i chcą Państwo uzyskać więcej informacji zachęcam do kontaktu:
tomasz.bieńTen adres pocztowy jest chroniony przed spamowaniem. Aby go zobaczyć, konieczne jest włączenie w przeglądarce obsługi JavaScript.


