


Monika Świontek
Instytut Chemii Organicznej, Politechnika Łódzka
strony wersji drukowanej: 6-17
strony wersji drukowanej: 6-17

Zarówno naturalne jak i syntetyczne peptydy są wysoce obiecującymi farmaceutykami, które mogą znaleźć szerokie zastosowanie w leczeniu różnorodnych chorób. Peptydy są zwykle wysoce selektywne i charakteryzują się wysoką skutecznością, a zatem jako farmaceutyki stosowane są zazwyczaj w małych ilościach. Niewątpliwą przewagą peptydów jako środków leczniczych jest fakt, że ich metabolizm prowadzi ostatecznie do nietoksycznych aminokwasów. Dla większości peptydów, właściwości te przekładają się na ich niską toksyczność oraz ograniczone ryzyko niepożądanych interakcji.
Wysoki potencjał farmaceutyczny peptydów dość często nie może zostać w pełni wykorzystany z uwagi na ich niską trwałość w warunkach in vivo. Zazwyczaj wiąże się to z szybkim rozkładem peptydów przez enzymy proteolityczne, choć należy pamiętać, że dużą wadą peptydów jako potencjalnych farmaceutyków jest również ich niewystarczająco efektywne wchłanianie przez błony śluzowe [1].
Na rynku dostępnych jest cały szereg farmaceutyków o budowie peptydowej. Najszerzej wykorzystywanymi są insulina, hormon uwalniającygonadotropinę, kalcytonina, enkefaliny, glukagon, inne analogi hormonów, inhibitory enzymów i szczepionki. W 2010 roku zatwierdzonych przez FDA do obrotu było ponad 50 leków peptydowych, dla których roczną sprzedaż globalną oceniono na około 1 mld USD. Przykładami tych leków są: cyklosporyna (Neoral®), octan gosereliny (Zoladex®), octan glatirameru (Copaxone®), octan leuprolidu (Lupron®) czy też octan oktreotydu (Sandostatin®). W tym samym roku w badaniach klinicznych było ponad 100 kolejnych leków peptydowych.
Początkowo, nieefektywne i kosztowne procesy wytwarzania peptydów w skali przemysłowej stanowiły znaczący czynnik hamujący rozwój terapeutyków peptydowych. Optymalizacje procesów wytwarzania sprawiły, że obecnie produkcja w skali ton rocznie enfuwirtydu (Fuzeon®), peptydu zbudowanego z 36 reszt aminokwasowych okazała się opłacalna ekonomicznie [2].
Efektywność wytwarzania peptydowych środków terapeutycznych zależy od wielu różnorodnych czynników takich jak: wielkość cząsteczki (długość łańcucha peptydowego), charakteru grup funkcyjnych w łańcuchach bocznych aminokwasów, wprowadzonych modyfikacji struktury, obecności nienaturalnych bloków budulcowych, rozpuszczalności, trwałości i skali syntezy. Stosowane są dwa główne podejścia do wytwarzania peptydowychśrodków terapeutycznych: synteza chemiczna oraz produkcja oparta o metody biotechnologiczne [3].
Chemiczna Synteza Peptydów jest szeroko stosowana zarówno w laboratoriach badawczych jak i w warunkach przemysłowych. Niewątpliwą przewagą chemicznej syntezy peptydów jest możliwość stosowania w trakcie syntezy praktycznie nieograniczonej liczby nienaturalnych aminokwasów, co jest trudne do osiągnięcia w procesach biotechnologicznych. Do najczęściej stosowanych metod chemicznej syntezy peptydów należy synteza w roztworze (SPS, ang. Solution-Phase Synthesis) oraz synteza w fazie stałej (SPPS, ang. Solid-Phase Peptide Synthesis). Oprócz tych dwóch głównych rozwiązań syntetycznych znanych jest wiele innych podejść obejmujących hybrydowe syntezy peptydów (HS, ang. hybride synthesis), chemoselektywną ligację (CL, ang. chemoselective ligation) i inne.
Główną zaletą SPS jest niewątpliwie aspekt ekonomiczny związany z brakiem konieczności stosowania dużych nadmiarów odczynników i substratów. Wadą natomiast jest konieczność izolowania i oczyszczania produktów pośrednich otrzymywanych w trakcie każdego kolejnego stadium syntezy peptydu. Tym samym technologie wytwarzania peptydów w roztworze są żmudne i czasochłonne. Pomimo tych ograniczeń farmaceutyki peptydowe zawierające od 3 reszt aminokwasowych (hormonuwalniający tyreotropinę) aż po peptydy zawierające ponad 30 reszt aminokwasowych (kalcytonina) otrzymywane były do badań klinicznych metodą syntezy w roztworze.
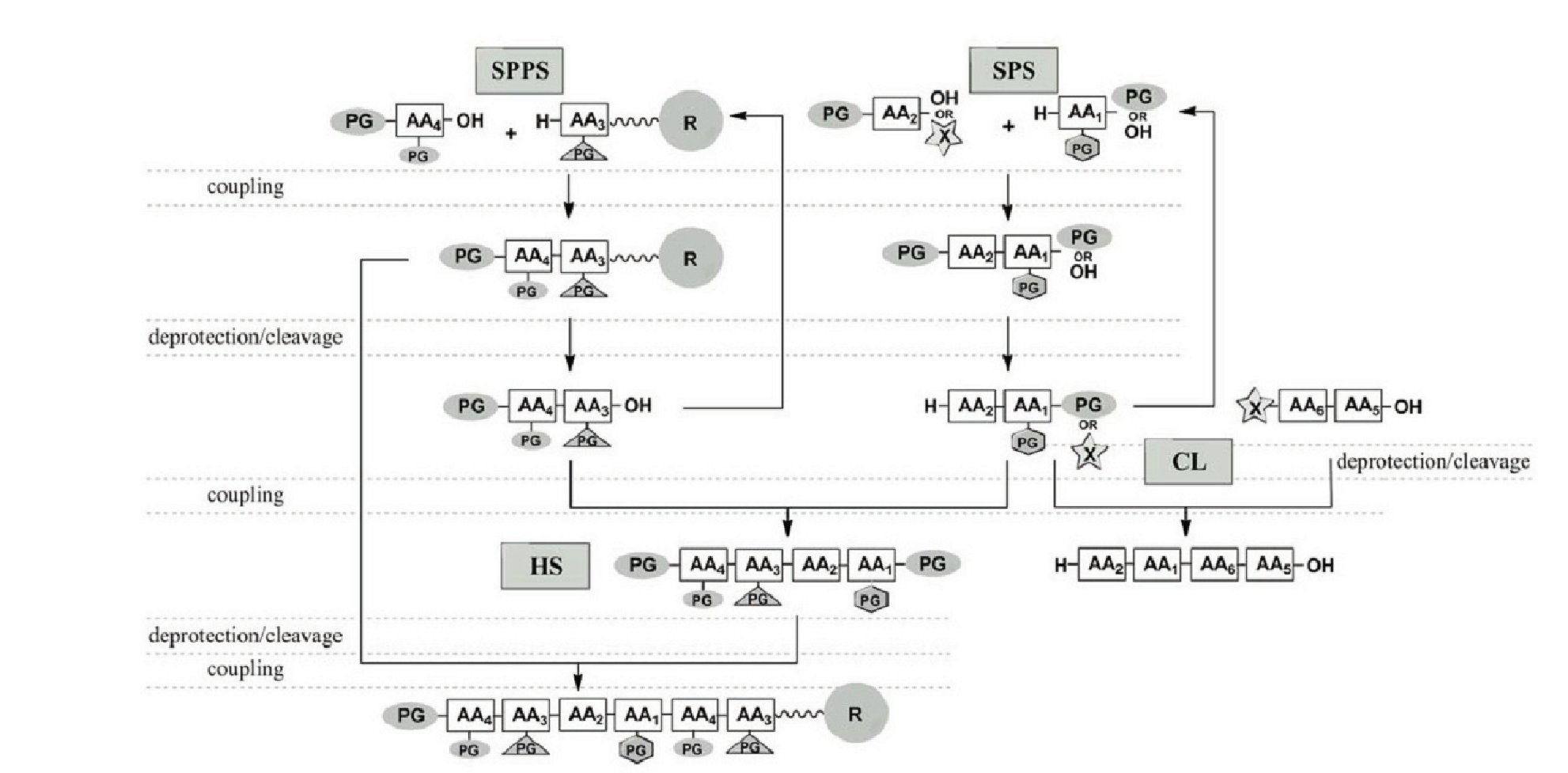
Rysunek 1. Chemiczne metody syntezy peptydów. Wszystkie metody obejmują powtarzające się cykle kondensacji oraz deprotekcji. PG: grupa ochronna; AA: aminokwas; X: ugrupowanie aktywujące grupę karboksylową; R: nośnik polimerowy [4].
Opracowanie przez Merrifielda metody syntezy na nośniku stałym znacząco uprościło procedury wytwarzania peptydów i w istotny sposób przyśpieszyło rozwój chemii peptydów. Metoda Merrifielda oparta jest na dobudowywaniu kolejnych blokowanych aminokwasów do pierwszego z nich zakotwiczonego na nierozpuszczalnej matrycy polimerowej. Podejście to umożliwiło automatyzację procesu wytwarzania peptydów. Ponadto metoda ta w porównaniu do syntez w roztworze jest szybsza i mniej pracochłonna.
Najważniejszym ograniczeniem stosowania metody SPPS na skalę przemysłową jest konieczność stosowania znacznego nadmiaru chronionych aminokwasów oraz odczynników kondensujących. Pomimo tej niedogodności wiele terapeutyków peptydowych otrzymanych jest tą metodą na skalę przemysłową. Najważniejszymi przykładami są zykonotyd (Prialt®), eksenatyd (Byetta®), pramlintyd (Symlin®, Amylin), degareliks (Firmagon®) i inne.
Do znaczącego postępu w rozwoju peptydowych środków leczniczych przyczyniło się również zastosowanie promieniowania mikrofalowego(microwave-assisted peptide synthesis). Jako sztandarowe przykłady zastosowania tej metody można wskazać syntezę Gramicydyny A oraz CSF114(Glc).
Odczynniki kondensujące w syntezie peptydów
Tworzenie wiązania amidowego pomiędzy grupą karboksylową oraz aminową aminokwasów jest kluczowym etapem syntezy peptydów. Pierwszym stadium kondensacji jest aktywacja funkcji karboksylowej. Proces ten jest często kluczowym etapem syntezy wiązania peptydowego.
Tylko dla niewielu odczynników aktywujących, produkt aktywacji może być stabilny i łatwy do izolowania. Zwykle produkt ten jest bezpośrednio konsumowany w kolejnym stadium reakcji. W drugim etapie, produkt pośredni jest atakowany przez odczynnik nukleofilowy (funkacja aminowa aminokwasu).
Znany jest cały szereg metod aktywacji funkcji karboksylowej [5] obejmujących tworzenie halogenków kwasowych (chlorki, fluorki), symetrycznych lub mieszanych bezwodników, estrów aktywnych oraz wykorzystywanych w tym celu różnorodnych odczynników kondensujących.
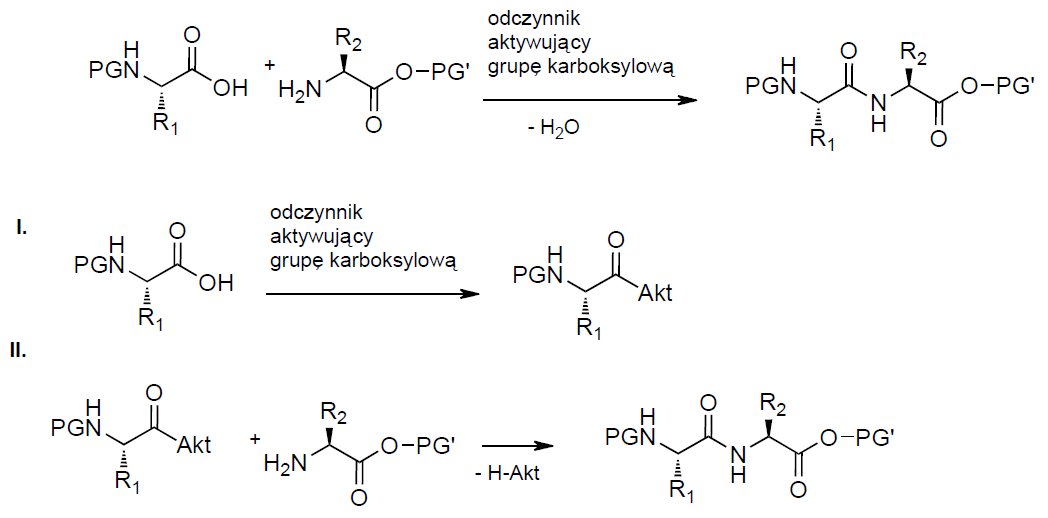
Rysunek 2. Synteza wiązania peptydowego.
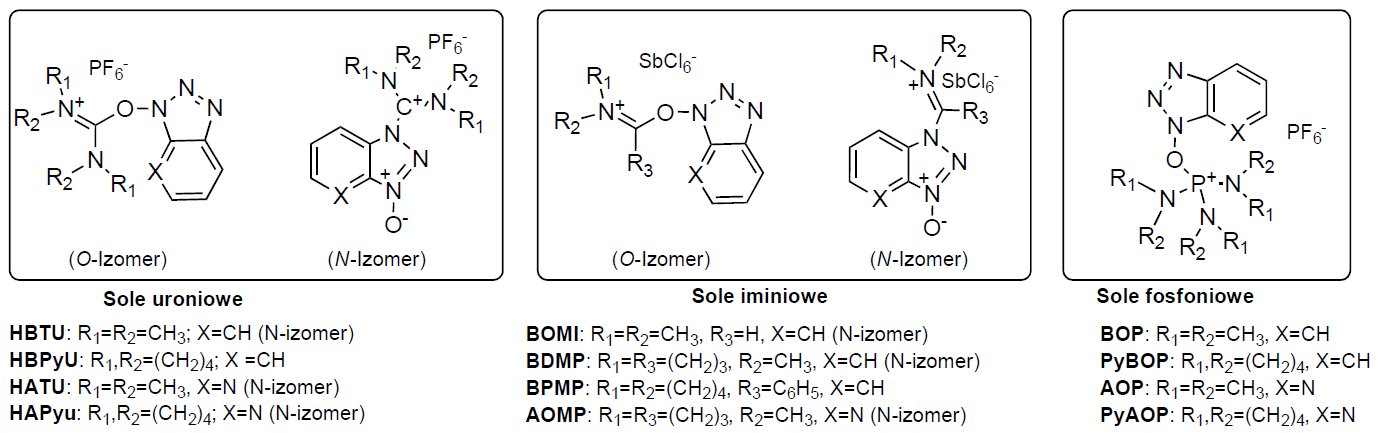
Rysunek 3. Struktury odczynników kondensujących wywodzących się z HOBt lub HOAt.
Przez wiele lat najczęściej stosowane były jako odczynniki kondensujące karbodiimidy (DCC, DIC, EDC x HCl) zarówno w syntezach z roztworze jak i syntezach w fazie stałej. Co prawda, karbodiimidy ciągle są wykorzystywane w syntezie peptydów, jednak w ostatnich latach coraz bardziej popularnymi odczynnikami kondensującymi stają się sole fosfoniowe, uroniowe (aminiowe) i iminiowe wywodzące się z HOBt (1-hydroksybenzotriazolu) lub HOAt (1-hydroksy-7-azabenzotriazolu).
Ograniczenie w przemysłowym stosowaniu odczynników kondensujących opartych o pochodne benzotriazolu spowodowane zostało ich podatnością do ulegania gwałtownemu rozkładowi zagrażającemu wybuchem.
W ostatnich latach popularnym odczynnikiem kondensującym stał się również COMU, którego efektywność jest porównywalna z HATU.

Rysunek 4. Struktura COMU i Oxyma Pure.
Wprowadzenie do cząsteczki (hydroksyimino)cyjanooctanu etylu (Oxyma Pure) w miejsce pochodnych benzotriazolu (HOBt lub HOAt) umożliwiło uzyskanie bezpieczniejszego odczynnika o lepszej rozpuszczalności i obniżonej alergennościw porównaniu do HBTU/TBTU lub HATU.
Należy jednak mieć świadomość, że żaden z odczynników kondensujących nie jest odczynnikiem uniwersalnym umożliwiającym realizację dowolnych celów syntetycznych.
Triazynowe Odczynniki Kondensujące
W wyniku badań prowadzonych w Instytucie Chemii Organicznej Politechniki Łódzkiej odkryty został nowy typ odczynników kondensujących. Otrzymywane in situ z 2-chloro-4,6-dimetoksy-1,3,5-triazyny (CDMT) i amin trzeciorzędowych chlorki N-triazynyloamoniowe pod działaniem kwasów karboksylowych tworzą estry o strukturze 2-acyloksy-1,3,5-triazyn a te z kolei reagując z aminami pierwszorzędowymi wyjątkowo łatwo tworząc odpowiednie amidy. Wysoka efektywność reakcji acylowania amin wpłynęła na zmianę mechanizmu tworzenia wiązania peptydowego, co spowodowało, że dla 2-acyloksy-4,6-dimetoksy-1,3,5-triazyn zaproponowano nazwę „estrów superaktywnych” [6].
Użyteczność triazynowych odczynników kondensujących wykazana została w licznych syntezach peptydów, estrów, bezwodników i amidów kwasów karboksylowych, antagonistów kwasu foliowego, β-laktamów, karetonoidów, fullerenów i innych związków [8].
Jednak obok doniesień dotyczących wysoce efektywnych aplikacji chlorków (4,6-dimetoksy-1,3,5-triazyn-2-ylo)-amoniowych jako odczynników kondensujących, w literaturze znaleźć możnarównież informacje o prowadzonych z ich udziałem reakcjach zakończonych mniej satysfakcjonującymi rezultatami [9].
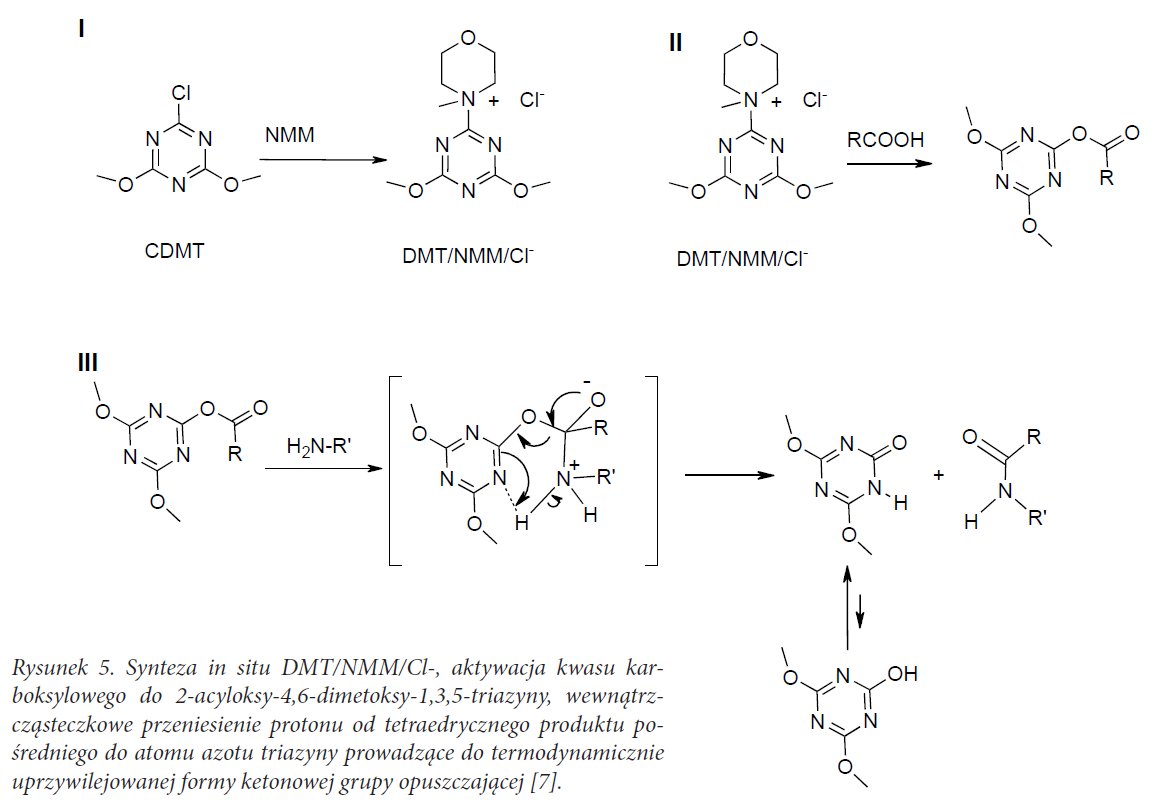
Rysunek 5. Synteza in situ DMT/NMM/Cl-, aktywacja kwasu karboksylowego do 2-acyloksy-4,6-dimetoksy-1,3,5-triazyny, wewnątrzcząsteczkowe przeniesienie protonu od tetraedrycznego produktu pośredniego do atomu azotu triazyny prowadzące do termodynamicznie uprzywilejowanej formy ketonowej grupy opuszczającej [7].
Okazało się, że chlorek 4-(4,6-dimetoksy-1,3,5-triazyn-2-ylo)-4-metylomorfoliniowy (DMT/NMM/Cl-), nie jest odczynnikiem dostatecznie trwałym w rozpuszczalnikach aprotonowych i łatwo ulega demetylacji, tworząc nieaktywną w procesie kondensacji 2,4-dimetoksy-6-morfolin-4-ylo-1,3,5-triazynę.
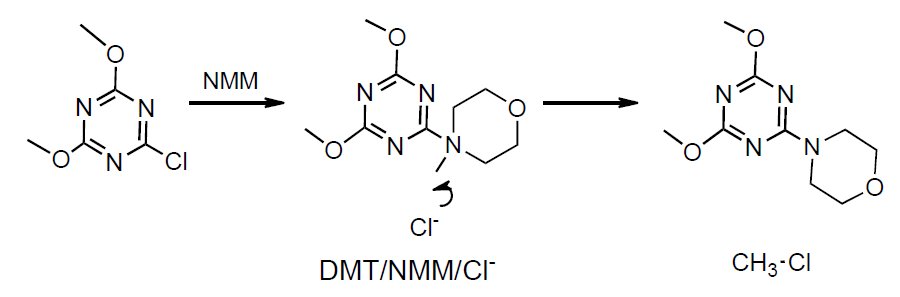
Rysunek 6. Synteza i dealkilacja czwartorzędowych chlorków N-triazynyloamoniowych.
Stało się jasne, że aplikacja czwartorzędowych chlorków N-triazynyloamoniowych jako odczynników kondensujących jest obarczona ryzykiemniepowodzenia z uwagi na ich nietrwałość i szereg reakcji ubocznych i jedynie stosowanie triazynowych odczynników kondensujących otrzymywanych in situ wydaje się być uzasadnione, ponieważ znacznie ogranicza ryzyko niepowodzenia reakcji kondensacji [10].
Rozwiązaniem problemu nietrwałości chlorków N-triazynyloamoniowych była wymiana nukleofilowego anionu chlorkowego na aniony o obniżonej nukleofilowości takie jak anion tetrafluoroboranowy, heksafluorofosforanowy, czy też pochodne kwasów sulfonowych [11].
Modułowa budowa triazynowych odczynników kondensujących otwiera dostęp do bibliotek różnorodnych soli N-triazynyloamoniowych o zróżnicowanych właściwościach.
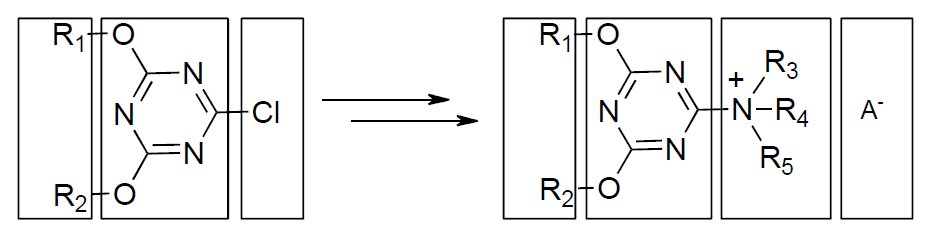
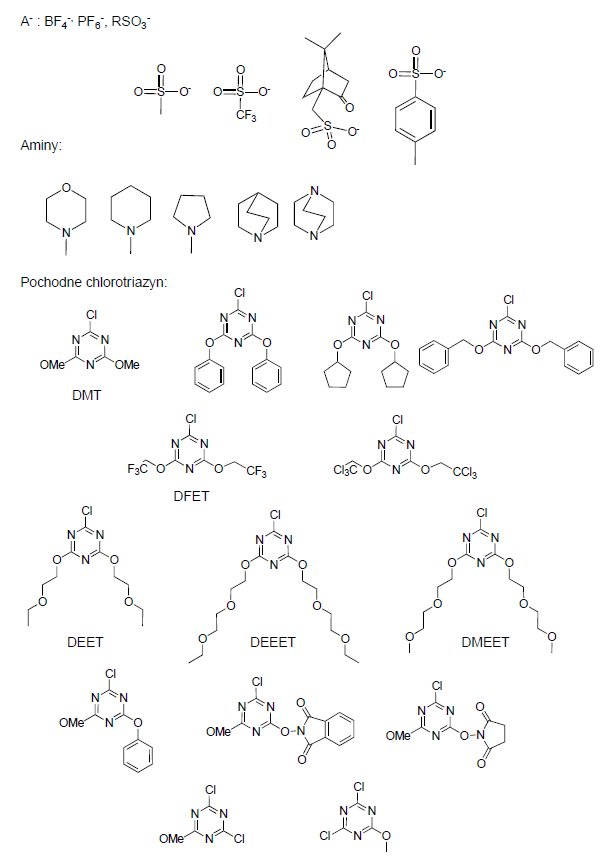
Rysunek 7. Modułowa budowa soli N-triazynyloamoniowych umożliwia syntezę zróżnicowanych strukturalnie triazynowych odczynników kondensujących.
Zarówno tertafluoroborany jak i sulfoniany N-triazynyloamoniowe są efektywnymi odczynnikami kondensującymi w syntezie peptydów, depsipeptydów, lipopeptydów jak i estrów zarówno w syntezach prowadzonych w roztworze jak i na fazie stałej. Swoją efektywnością dorównują, a nawet przewyższają najlepsze znane obecnie odczynniki wywodzące się z N-hydroksybenzotriazolu lub N-hydroksy-7-azabenzotriazolu.
Za szczególnie cenną uznać należy wyjątkowo wysoką efektywność triazynowych odczynników kondensujących w tzw „trudnych” kondensacjach,zachodzących z udziałem sterycznie zatłoczonych substratów oraz skuteczność w acylowaniu substratów o obniżonej nukleofilowości oraz peptydów podatnych na agregację.
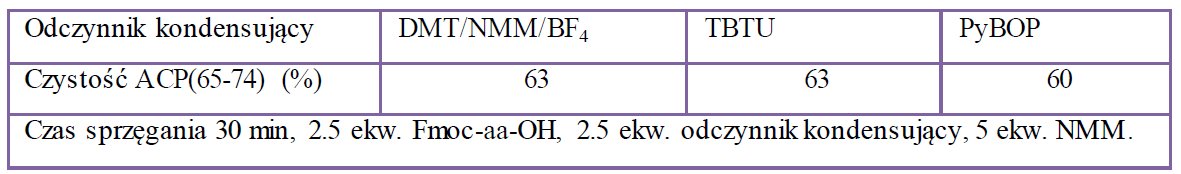
Rekomendowanym do testowania efektywności odczynników kondensujących, jest fragment 65-74 białka ACP. W badaniach porównawczych, w których jako odczynniki zastosowano TBTU, PyBOP oraz DMT/NMM/BF4 stwierdzono, że triazynowy odczynnik kondensujący umożliwia otrzymanie finalnego peptydu z porównywalną czystością [11a].
Tabela 1. Zestawienie wyników automatycznej syntezy fragmentu 65-74 białka ACP.
Tabela 1. Zestawienie wyników automatycznej syntezy fragmentu 65-74 białka ACP.
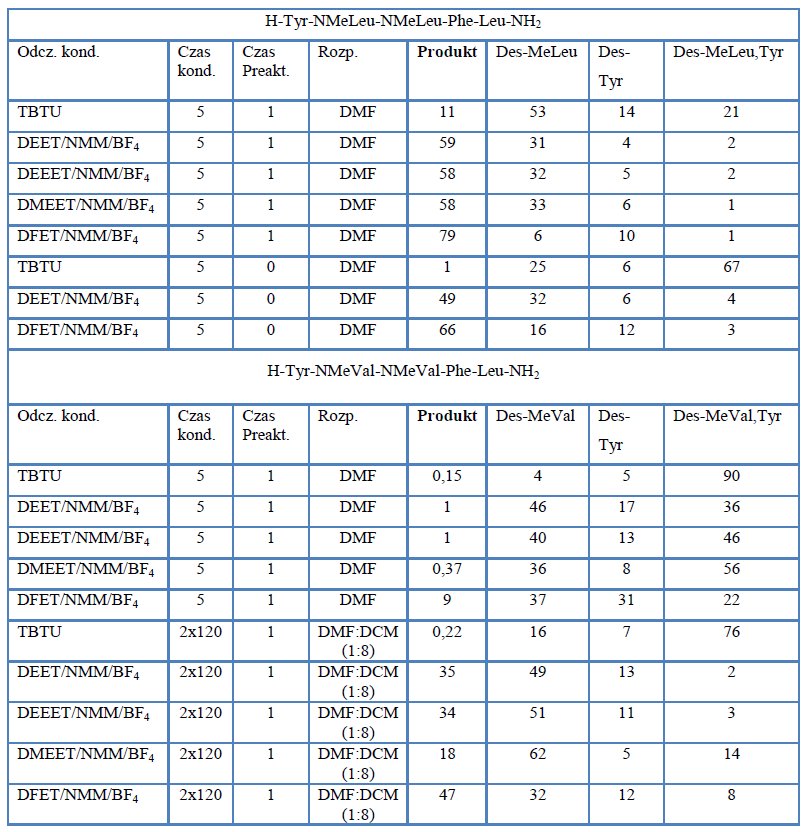
Porównano również efektywność triazynowych odczynników kondensujących w syntezie analogów enkefaliny modyfikowanych resztami N-metylowanych aminokwasów. Jedynie w syntezie Tyr-NMeGly-NMEGly-Phe-Leu nie obserwowano różnicy w efektywnościTBTU oraz tetrafluoroboranów N-triazynyloamoniowych. Jednak w przypadku syntezy analogów zawierających reszty N-metyloleucyny oraz N-metylowaliny widoczna była ogromna przewaga triazynowych odczynników kondensujących [12].
Tabela 2. Zestawienie wyników syntezy analogów enkefaliny.
Tabela 2. Zestawienie wyników syntezy analogów enkefaliny.
Zarówno tetrafluoroboran 4-(4,6-dimetoksy-1,3,5-triazyn-2-ylo)-metylomorfoliniowy (DMT/NMM/BF4) jak i p-toluenosulfonian 4-(4,6-dimetoksy-1,3,5-triazyn-2-ylo)-metylomorfoliniowy (DMT/NMM/TsO) z powodzeniem zostały również zastosowane w syntezach wysoce podatnych na agregację rdzeni amyloidogennych polipeptydów/białek ulegających samoorganizacji. Agregujące fragmenty ludzkiej amyliny, insuliny, kalcytoniny oraz β-amyloidu otrzymane zostały z wysokimi wydajnościami oraz czystościamisurowych produktów przekraczającymi 85%.
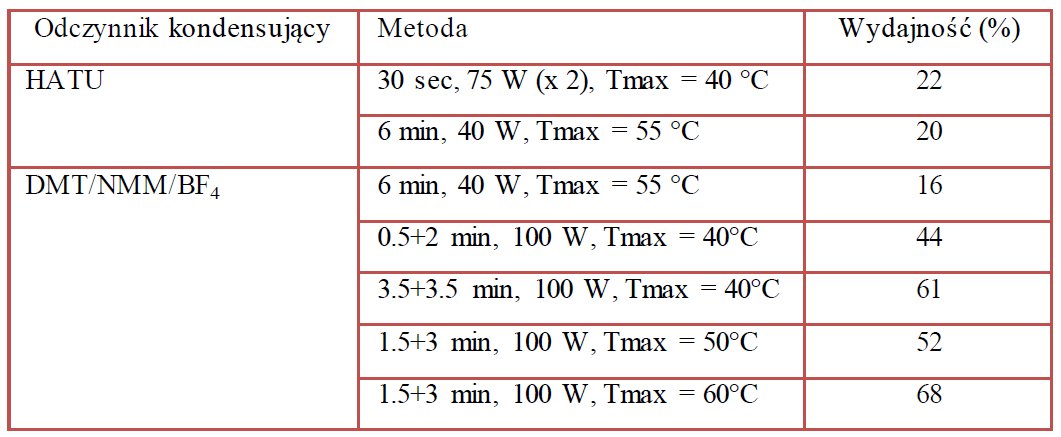
Okazało się również, że wyselekcjonowane triazynowe odczynniki kondensujące mogą być z powodzeniem stosowane we wspomaganych promieniowaniem mikrofalowym syntezach modyfikowanych aminokwasów jak i trudnych sekwencjach peptydowych. We wspomaganej promieniowaniem mikrofalowym syntezie Fmoc-Asn-(Glc)-OtBu w roztworze DMT/NMM/BF4 znacznie przewyższał swoją efektywnością HATU [13].
Tabela 3. Zestawienie wyników wspomaganej promieniowaniem mikrofalowym syntezy Fmoc-Asn(Glc)-OtBu.
Tabela 3. Zestawienie wyników wspomaganej promieniowaniem mikrofalowym syntezy Fmoc-Asn(Glc)-OtBu.
Z kolei DMT/NMM/TsO okazał się bardzo efektywnym odczynnikiem kondensującym we wspomaganych promieniowaniem mikrofalowym syntezach w fazie stałej glikozylowanych peptydów:
- (292-300) gp120 H-N(Glc)ESVAIN(Glc)CT-OH,
- CSF114(Glc) H-TPRVERN(Glc)GHSVFLAPYGWMVK-OH,
- hMOG H-KN(Glc)ATGMEVGWYRPPFSRVVHL-OH,
- FAN(635-655)Glc H-GITVSRN(Glc)GSSVFTTSQDSTLK-OH.
We wszystkich przypadkach czystości surowych produktów przekraczały 83%.
Triazynowe odczynniki kondensujące okazały się być również bardzo efektywne we wspomaganej promieniowaniem mikrofalowym syntezie inhibitorów agregacji polipeptydów/białek, które w swoich strukturach zawierają reszty N-metylowanych aminokwasów.
Zastosowanie promieniowania mikrofalowego nie tylko skraca czas syntezy, ale również umożliwia otrzymywanie produktów o wysokiej czystości.
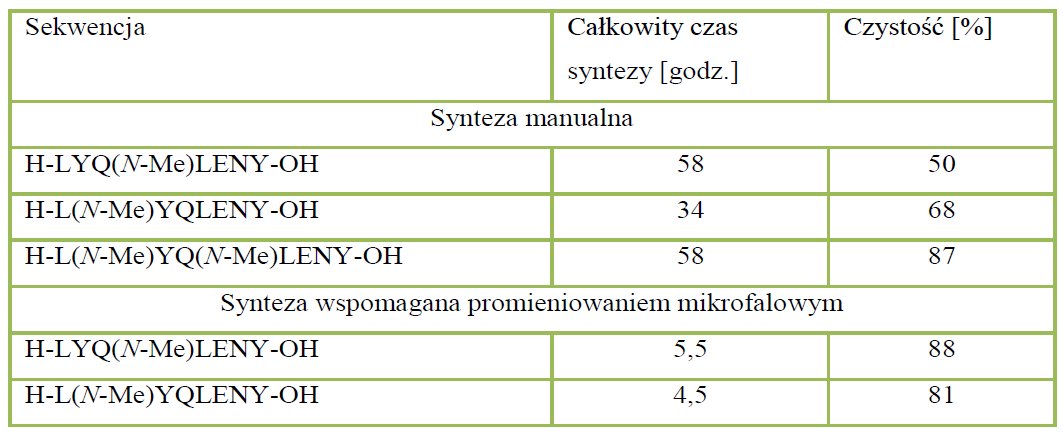
Tabela 4. Zestawienie wyników syntezy modyfikowanych za pomocą reszt N-metylowanych aminokwasów analogów amyloidogennego fragmentu ludzkiej insuliny.
Tabela 4. Zestawienie wyników syntezy modyfikowanych za pomocą reszt N-metylowanych aminokwasów analogów amyloidogennego fragmentu ludzkiej insuliny.
Podsumowanie
Naturalne czynniki regulujące homeostazę, do jakich należą peptydy, stają się coraz łatwiejdostępnymi w skali umożliwiającej ich szerokie wprowadzenie do lecznictwa. Postęp w tym zakresie dokonany został zarówno poprzez intensywnie prowadzone poszukiwania nowych, wyrafinowanych struktur peptydowych jak i poprzez udoskonalenie metod syntezy wiązania peptydowego umożliwiających ich wydajną syntezę nawet w przypadkach najtrudniejszych sekwencji peptydowych i niereaktywnych bloków budulcowych.
Literatura:
[1] Reichert, J.; Pechon, P.; Tartat, A.; Dunn, M.K. Development trends for peptide therapeutics: A comprehensive quantitative analysis of peptide therapeutics in clinical development.; Peptide Therapeutics Foundation: 2010.
[2] Bray, B.L. Nat. Rev. Drug Discov., 2003, 2, 587-593.
[3] Thayer, A.M. Chem. Eng. News, 2011, 89, 21-25.
[4] Goodwin, D.; Simerska, P.; Toth, I. Curr. Med. Chem., 2012, 19, 4451-4461.[5] a) Valeur, E.; Bradley, M., Chem.Soc.Rev., 2009, 38, 606–631; b) Subirós-Funosas, R.; Khattab, S.N.; Nieto-Rodríguez, L.; El-Faham, A.; Albericio. F., Aldrichimica Acta, 2013, 1, 46; c) Al-Warhia, T.I.; Al-Hazimib, H.M.A.; El-Faham, A., J. Saudi Chem. Soc., 2012, 16, 97–116; d) El-Faham, A.; Albericio, F., Chem. Rev., 2011, 111, 6557–6602.
[6] Kaminski Z.J. Int. J. Peptide Protein Res., 1994, 43, 312-319.
[7] a) Kamiński, Z.J.; Paneth, P.; Rudziński, J. J. Org. Chem., 1998, 63, 4248-4255; b) Kamiński, Z.J.; Paneth, P.; O’Leary, M. J. Org. Chem., 1991, 56, 5716-5719.
[8] Kamiński, Z.J., Biopolymers, 2000, 55, 140-165.
[9] Sabatino, G.; Mulinacci, B.; Alcaro, M.C.; Chelli, M.; Rovero, P.; Papini, A.M., Lett. Pept. Sci., 2002, 9, 119- 123.
[10] a) Kolesińska, B., Kamiński, Z.J. Polish J. Chem., 2008, 82, 2115–2123; b) Kolesińska, B., Kamiński, Z.J., Tetrahedron, 2009, 65, 3573–3576.


