


Marta Bauer
Gdański Uniwersytet Medyczny
strony wersji drukowanej: 57-62
strony wersji drukowanej: 57-62
Problemy związane z metabolizmem aminokwasów wynikają przede wszystkim z braku lub niedoboru określonego enzymu szlaku katabolizmu białek. Najczęściej zaburzenia te dotyczą jednego aminokwasu, choć nie jest to regułą. Defekty enzymatyczne powodują, że aminokwasy, które nie uległy metabolizmowi występują w nadmiarze i kumulują się w ważnych narządach, tj. mózgu czy wątrobie, stanowiąc bezpośrednie zagrożenie życia. Substraty nie są wykorzystywane przez organizm, co w konsekwencji prowadzi do deficytu innych substancji potrzebnych do prawidłowego funkcjonowania. Udowodnione jest, że w odpowiedzi na tego typu sytuację stresową organizm ludzki potrafi wytworzyć boczne szlaki metaboliczne mające na celu zrekompensowanie strat. Możliwe jest również powstanie nowych szlaków z udziałem substancji, których obecność nie jest stwierdzana w okresie homeostazy. Dotychczas opisano ok. 70 rodzajów defektów przemian aminokwasów określanych mianem aminoacidopatii. Zdecydowana większość to zaburzenia wrodzone, których przyczyną są zmiany genetyczne dziedziczone w sposób autosomalny recesywny. W przypadku występowania pierwotnego defektu enzymu niezwykle istotne jest jak najszybsze rozpoznanie i wdrożenie odpowiedniego leczenia. Jest to trudne ze względu na mylące i niespecyficzne objawy. Sprawne postępowanie przy podejrzeniu choroby metabolicznej daje większe szanse na prawidłowy rozwój dziecka.
Duże znaczenie ma również program badań przesiewowych noworodków, który obejmuje fenyloketonurię, chorobę syropu klonowego i homocystynurię. Wymienione schorzenia są najczęstsze i zostaną dokładniej omówione w dalszej części.
PODSTAWOWA DIAGNOSTYKA LABORATORYJNA
Testy w kierunku występowania wrodzonych zaburzeń przemian aminokwasów polegają przede wszystkim na ilościowym określeniu obecności aminokwasów w osoczu pacjenta, badaniach molekularnych i ustaleniu aktywności enzymu. Ogólnie, potwierdzenie nieprawidłowego metabolizmu aminokwasów opiera się na:
- wykonaniu mikrobiologicznego testu hamowania Guthriego (do 5. dnia życia dziecka);
- określeniu poziomu produktów przemian białek metodami chromatografii;
- stwierdzeniu obecności mutacji w genie odpowiedzialnym za działanie określonego enzymu.
Test hamowania uznawany jest za badanie przesiewowe. Wykorzystuje się w nim szczep Bacillus subtilis, a interpretacja polega na obserwacji strefy wzrostu drobnoustroju, która jest proporcjonalna do ilości oznaczanego aminokwasu. We wszystkich wyżej wspomnianych badaniach wykorzystywana jest próbka krwi włośniczkowej pobrana z pięty dziecka i naniesiona na bibułkę filtracyjną. Analiza przeprowadzana jest po wysuszeniu materiału.
Inne, bardziej zaawansowane metody diagnostyczne zostaną omówione w dalszej części artykułu.
FENYLOKETONURIA
Fenyloketonuria jest najczęściej występującym zaburzeniem metabolizmu aminokwasów spośród poznanych. Częstotliwość z jaką rodzą się dzieci z fenyloketonurią wynosi ok. 1:10000. Podstawą tej choroby jest brak lub niedostateczna aktywność hydroksylazy fenyloalaniny- enzymu produkowanego w wątrobie i odpowiedzialnego za hydroksylację fenyloalaniny do tyrozyny. W badaniach laboratoryjnych obserwowany jest wzrost stężenia fenyloalaniny w osoczu, której ilość wpływa na metabolizm tryptofanu. Zahamowanie jego transportu z jelita cienkiego odbywa się na zasadzie konkurencji o wiązanie do miejsca transportowego, czego wynikiem jest spadek produkcji serotoniny.
Cechą charakterystyczną fenyloketonurii jest brak objawów klinicznych w pierwszych miesiącach życia noworodka. Dziecko rodzi się zdrowe. Pierwsze symptomy choroby pojawiają się ok. 3 miesiąca. Należą do nich:
- zaburzenia rozwoju motorycznego i umysłowego;
- mikroencefalopatia;
- nieprawidłowa postawa ciała i hipotonia mięśni;
- wymioty z towarzyszącymi drgawkami;
- „mysi” zapach moczu i potu dziecka.
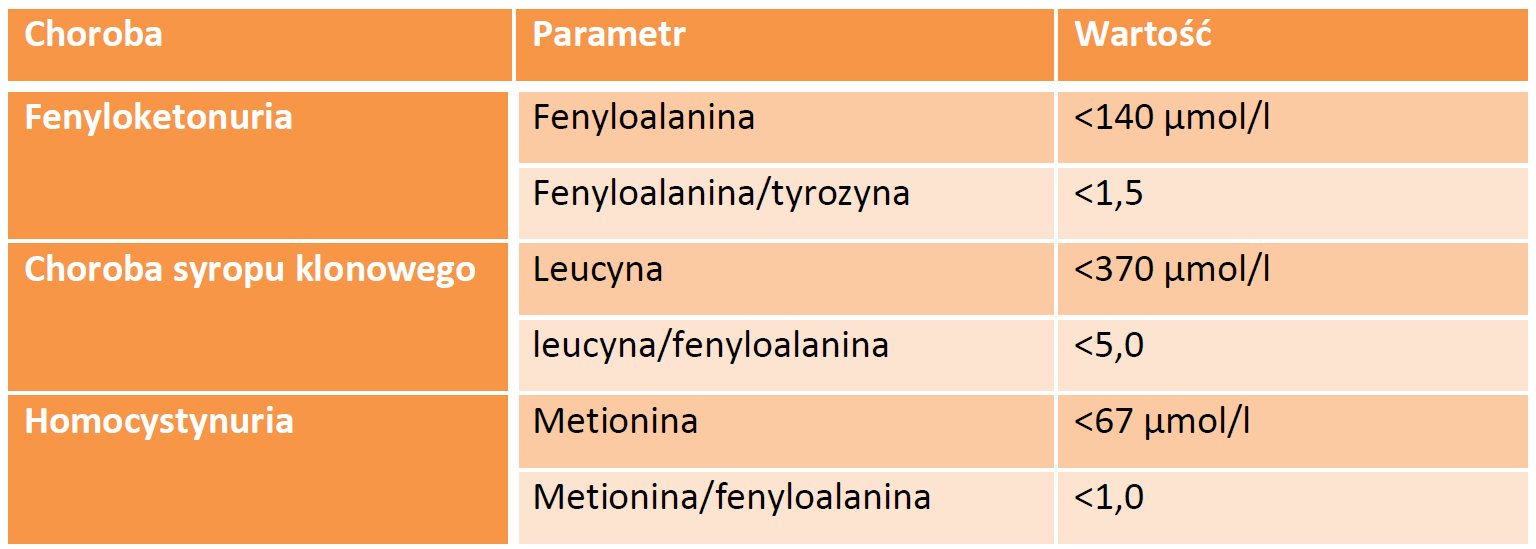
Tab. 1. Wartości referencyjne parametrów ocenianych w rozpoznawaniu zaburzeń przemian aminokwasów.
Podstawą leczenia dzieci chorych na fenyloketonurię jest zastosowanie diety eliminacyjnej redukującej podaż fenyloalaniny. Istotne jest, aby zmieniony jadłospis wprowadzić jeszcze przed 3 miesiącem życia - tylko w ten sposób można liczyć na skuteczność terapii.
CHOROBA SYROPU KLONOWEGO
W literaturze zaburzenie to nazywane jest fachowo ketoacydurią aminokwasów rozgałęzionych. Jest to choroba rzadka, częstość jej występowania szacowana jest na 1:200000. Podłożem choroby syropu klonowego jest defekt w enzymach odpowiedzialnych za przemiany aminokwasów rozgałęzionych (np. leucyny, waliny), co skutkuje ich wysokim stężeniem we krwi i moczu. W przypadku braku leczenia następuje intoksykacja prowadząca do zgonu dziecka lub wystąpienia trwałych uszkodzeń psychoruchowych.
W odróżnieniu od fenyloketonurii objawy kliniczne choroby syropu klonowego pojawiają się już ok. 5. dnia życia noworodka. Jednym z najbardziej charakterystycznych symptomów jest zapach moczu przypominający zapach karmelu lub syropu klonowego- stąd potoczna nazwa choroby. Poza tym, grupa objawów obejmuje:
- problemy z karmieniem;
- tachypnoe;
- skurcze mięśni;
- apatię;
- epizody padaczkowe.
W przebiegu choroby syropu klonowego możliwe jest wystąpienie hiperglikemii i kwasicy metabolicznej spowodowanej obecnością α-ketokwasów.
Leczenie opiera się na ograniczeniu spożycia aminokwasów rozgałęzionych do ilości niezbędnej dla prawidłowego rozwoju. W przypadku wystąpienia kwasicy niezbędna jest hemodializa. Metodą leczenia, która osiąga najlepsze skutki jest przeszczep wątroby.
HOMOCYSTYNURIA
Choroba spowodowana nieprawidłową przemianą metioniny i homocysteiny występująca z częstością 1:200000. Wywołana jest przez niedostateczną aktywność enzymów zależnych od witamin z grupy B: β-syntazy cystationinowej lub syntazy/reduktazy metioninowej. Tak jak we wszystkich omawianych zaburzeniach dochodzi do akumulacji aminokwasów, co wywołuje szereg objawów:
- wysoki wzrost;
- skoliozę;
- osteoporozę;
- niedorozwój umysłowy;
- deformacje klatki piersiowej;
- drgawki;
- zmiany miażdżycowe w tętnicach;
- zmiany w soczewce oka.
Poza leczeniem opartym na redukcji spożycia metioniny skuteczna jest również suplementacja pirydoksyną.
INNE ZABURZENIA METABOLIZMU AMINOKWASÓW
Poza wymienionymi wyżej istnieje szereg innych schorzeń, których podstawą są defekty przemian aminokwasów, jednak ze względu na nieliczne przypadki występowania często są pomijane.
Alkaptonuria (Ochronoza) przebiega ze spichrzaniem w tkankach kwasu homogentyzynowego, co prowadzi do zmian zwyrodnieniowych w stawach. Inną chorobą wynikającą z nieprawidłowego metabolizmu aminokwasów jest tyrozynemia: typ I i II. W pierwszym przypadku mamy do czynienia z brakiem hydroksylazy fumaryloacetooctanu. Typ II związany jest z deficytem transaminazy tyrozyny. Problemy w przemianach aminokwasów nie muszą dotyczyć enzymu biorącego udział w ich katabolizmie. Istnieją jednostki chorobowe, w których występują zaburzenia w transporcie aminokwasów, jak np. choroba Hartnupów lub zespół Fanconiego.
METODY STOSOWANE W ANALIZIE AMINOKWASÓW
Za najbardziej optymalną technikę analizy składu aminokwasów uważa się taką, która charakteryzuje się wysoką szybkością oznaczania, prostotą, odpowiednia czułością i powtarzalnością. Oczywiście ważną cechą jest koszt analiz. Jedną z najprostszych, a jednocześnie najczęściej wykorzystywanych technik w rozpoznawaniu zaburzeń metabolizmu aminokwasów jest chromatografia cienkowarstwowa. Metoda służy głównie jako szybki i tani test przesiewowy. Bardziej zaawansowane technologie opierają się na zautomatyzowaniu chromatografii jonowymiennej. Chętnie wykorzystywana jest również chromatografia cieczowa z odwróconym układem faz. Wspomniane technologie uważane są najbardziej optymalne ze względu na spełnianie oczekiwań metody idealnej. Inaczej jest w przypadku chromatografii gazowej, której zastosowanie jest bardzo ograniczone ze względu na konieczność przeprowadzania analitu w stan gazowy. Dużą popularnością w laboratoriach diagnostycznych cieszy się także tandemowa spektrometria masowa (MS/MS). Technika ta opiera się na wykorzystaniu 2 spektrometrów masowych połączonych komorą. W pierwszej części następuje jonizacja molekuł wprowadzonej próbki. Następnie jony rozbijane są na małe fragmenty i przeprowadzane są do drugiego urządzenia, gdzie cząstki segregowane są pod względem wielkości. Metoda MS/MS jest szybka i pozwala na rozpoznawanie kilku zaburzeń jednocześnie. Szczególne zastosowanie znalazła w diagnostyce fenylokteonurii.
Opisując metody diagnostyczne warto wspomnieć o technikach uważanych za innowacyjne. W ustalaniu składu aminokwasowego znalazła zastosowanie chromatografia na kolumnach kapilarnych, która z powodzeniem może być łączona z elektrochromatografią. Niewątpliwą zaletą tej metody jest rozdział, który w kapilarach zajmuje kilka minut - dotyczy to także dużej ilości aminokwasów.
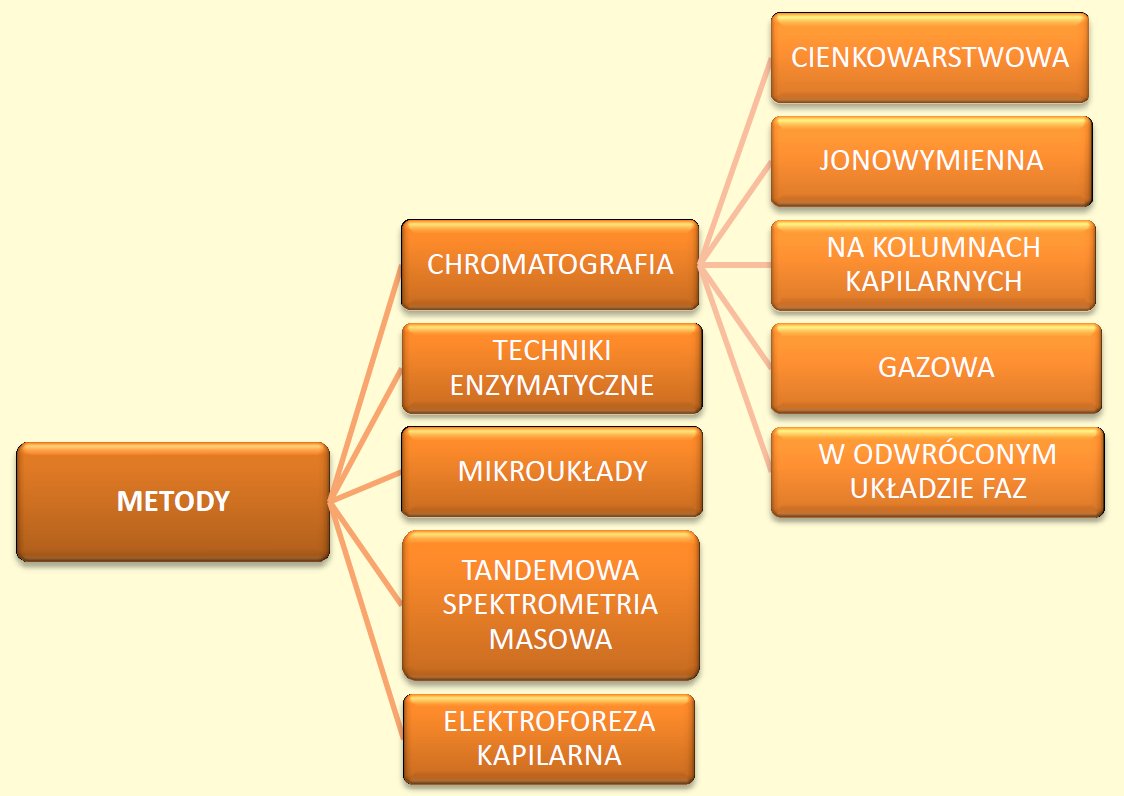
Ryc. 1. Techniki analityczne wykorzystywane w analizie składu aminokwasów.
Kolejną techniką jest elektroforeza kapilarna podczas której aminokwasy rozdzielane są pod wpływem wysokiego napięcia, a rozdział przeprowadzany jest na podstawie ładunków jakie posiadają oznaczane substancje. Mówiąc o metodach diagnostyki zaburzeń aminokwasów warto wspomnieć o technologiach wykorzystujących enzymy. Zasada oznaczania aminokwasów polega na zastosowaniu elektrody opłaszczonej oksydazą aminokwasów, którą umieszcza się w roztworze analizowanej próbki. Wynik określany jest na podstawie ilości produktów utlenienia. Obecnie dużo uwagi poświęca się na rozwój metod opartych na mikroukładach. Ustalanie składu aminokwasów polega na zastosowaniu mikrokanałów, w których rozdział zachodzi na zasadzie elektroosmozy. W laboratoriach powstają prototypy tego typu układów, a dotychczasowe wyniki dają szanse na dynamiczny postęp w tym temacie.
PODSUMOWANIE
Mimo, że opisane powyżej wrodzone zaburzenia metabolizmu aminokwasów nie występują często, warto zwracać uwagę na ich diagnostykę, ponieważ warunkuje ona możliwość uratowania życia nowonarodzonego dziecka i zapewnienia mu prawidłowego rozwoju. Istotne jest przeprowadzanie badań przesiewowych, a także zbieranie dokładnego wywiadu lekarskiego w celu ustalenia ewentualnego obciążenia genetycznego. Badania molekularne pozwalające ustalić obecność defektu enzymatycznego mogą być wykonywane już w okresie życia płodowego, dzięki czemu lekarze są w stanie dobrać odpowiedni sposób leczenia. Problemem są jednak częste błędy diagnostyczne i nieprawidłowe rozpoznanie. Lekarze pierwszego kontaktu często nie biorą pod uwagę możliwości wystąpienia zaburzeń metabolicznych, dlatego ważne jest aby nagłaśniać ten temat i o nim mówić.
PIŚMIENNICTWO:
1. Banta-Wright S. A., Steiner R. D., Tandem mass spectrometry in newborn screening. A primer for neonatal and perinatal nurses. J Perinat Neonat Nurs, 18, 41-58 (2004).
2. Dembińska-Kieć A., Naskalski J. W., Diagnostyka laboratoryjna z elementami biochemii klinicznej. Elsevier Urban & Partner (2010).
3. Marcdante K. J., Kliegman R. M., Jenson H. B., Behrman R. E., Nelson Pediatria t. I. Elsevier Urban & Partner (2012).
4. Neumeister B., Besenthal I., Bohm B. O., Diagnostyka laboratoryjna. Poradnik kliniczny. Elsevier Urban & Partner (2013).
5. Zahou E., Jornvall H., Bergman T., Amino acid analysis by capillary electrophoresis after phenylthiocarbomylation. Anal Biochem, 15, 115-122 (2000).


