


Damian Neubauer
Gdański Uniwersytet Medyczny
Peptydy ze względu na bogactwo zastosowań oraz efektów biologicznych, które mogą wywoływać, są obiektem szczególnego zainteresowania środowisk naukowych oraz przemysłu, w tym farmaceutycznego. Bez ich udziału niemożliwym byłoby funkcjonowanie organizmów żywych, czego dowodem są np. hormony. Związki peptydowe znalazły zastosowanie też jako antybiotyki. Przykładem jest kolistyna będąca lekiem stosowanym w leczeniu zakażeń wywołanych wieloopornymi bakteriami Gram-ujemnymi, oraz oktreotyd będący syntetycznym analogiem somatostatyny stosowanym u pacjentów z akromegalią.1 Ponadto analog oktreotydu zawierający radionuklid (np. 111In) jest stosowany w celowanej terapii przeciwnowotworowej.2
W celu wykorzystania peptydów na skalę przemysłową stosowane są metody syntezy chemicznej, izolacji z materiału biologicznego, biosyntezy w komórkach organizmów modyfikowanych genetycznie, a także kombinacje powyższych metod. Pierwotnie syntezę chemiczną prowadzono w roztworze. Metoda ta jest wciąż stosowana ze względu na korzystny bilans ekonomiczny. Obecnie chemiczna synteza peptydów, szczególnie ta prowadzona w skali laboratoryjnej, opiera się głównie na syntezie na nośniku stałym (SPPS, ang. Solid-phase peptide synthesis). Metoda została zaproponowana w roku 1963 przez R.B. Merrifield’a, późniejszego laureata Nagrody Nobla. Z biegiem czasu koncepcja syntezy w fazie stałej została rozszerzona i zaaplikowana w syntezie innych grup związków, na przykład oligonukleotydów. SPPS zdecydowanie uprościła procedurę otrzymywania peptydów, co zaowocowało dynamicznym jej upowszechnieniem. Najczęściej stosowana w otrzymywaniu syntetycznych peptydów jest tzw. chemia Fmoc (9 fluorenylometoksykarbonyl), gdzie do tymczasowej ochrony grupy Nα-aminowej wykorzystuje się ugrupowanie zasadolabilne. Synteza na nośniku stałym ma charakter wieloetapowy, co niesie za sobą konieczność kontrolowania poszczególnych procesów. Wpływa to w sposób istotny na wydajność syntezy, która w przypadku SPPS jest mniejsza (często otrzymywana jest mieszanina związków, których rozdzielenie jest problematyczne). Problematyka syntezy peptydów poza zagadnieniem wydajności obejmuje także czystość optyczną związku, który składa się z reszt aminokwasowych charakteryzujących się konkretną konfiguracją przestrzenną, a jej zachowanie stanowi zwykle podstawę uzyskania pożądanych efektów w układzie biologicznym. Z tych właśnie względów istotne jest opracowywanie nowych, coraz doskonalszych środków sprzęgających, hamujących proces racemizacji, czy też optymalizacja warunków syntezy i jej przyspieszenie. Nie bez znaczenia jest też aspekt ekologiczny syntezy, nad którym pracują liczne ośrodki badawcze proponując mniej szkodliwe zamienniki i opracowując niekonwencjonalne warunki prowadzenia reakcji np. w środowisku wodnym.3,4 Wspomniany wieloetapowy charakter syntezy oraz jej spopularyzowanie były niewątpliwie przyczyną do rozpoczęcia prac nad automatyzacją procesu. Pewnym ułatwieniem w realizacji tego założenia jest powtarzalność poszczególnych etapów: sprzęgania, przemywania żywicy i deprotekcji grupy aminowej.
W artykule wskazano na wybrane problemy występujące podczas chemicznej syntezy peptydów i podano przykłady rozwiązań. Przedstawiono także najnowsze osiągnięcia w dziedzinie syntezy, na które składają się nowe typy reagentów oraz rozwiązania zastosowane w automatycznych syntezatorach peptydów.
Automatyczne syntezatory peptydów, do roboty!
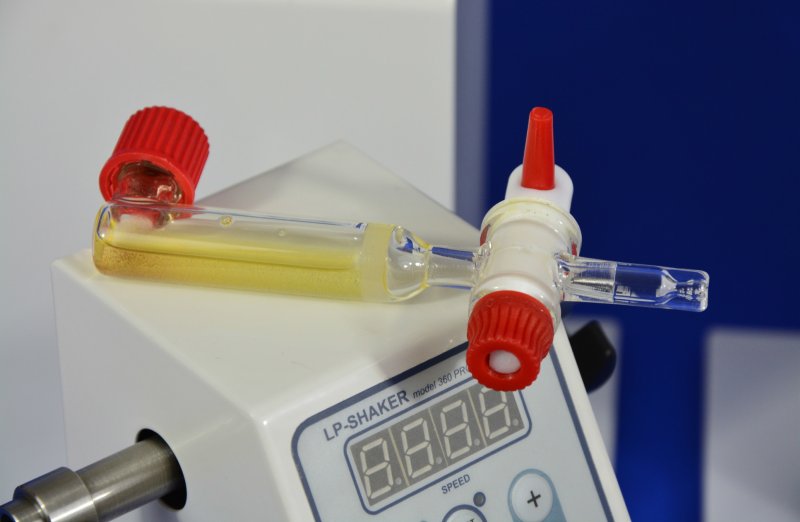
Synteza peptydów prowadzona być może w sposób manualny, pół-automatyczny, jak i automatyczny. Do pierwszego z wymienionych sposobów wykorzystywane są proste naczynia szklane ze spiekiem szklanym lub porowatym filtrem (rys. 1), zaś mieszanina reakcyjna wytrząsana jest zwykle ruchem obrotowym, orbitalnym, lub posuwisto-zwrotnym na odpowiedniej wytrząsarce (przykłady wytrząsarek „peptydowych” oraz specjalistycznego szkła można porównać na stronie www.peptideweb.com).
Rys. 1. Naczynie do syntezy manualnej.5

Rys. 2. Wytrząsarka LP-shaker 360 AMP.6
Pierwsze automatyczne syntezatory peptydów zostały skonstruowane w latach 60. ubiegłego wieku. Po dziś dzień opierają się one na układzie podajników reagentów kierowanych do naczynia reakcyjnego zawierającego peptydylo-żywicę. Z biegiem czasu systemy ulegały udoskonaleniom opierającym się głównie na optymalizacji mieszania, zapobieganiu kontaminacji odczynnikami z uprzednich etapów syntezy (cofanie reagentów), próbie bieżącej oceny wydajności reakcji sprzęgania i deprotekcji w celu przedsięwzięcia odpowiednich kroków w razie niezadowalającego przebiegu powyższych procesów, wspomaganiu reakcji za pomocą podwyższonej temperatury oraz ujednoliceniu jej rozkładu wewnątrz naczynia reakcyjnego, także poprzez zmianę sposobu dostarczania energii do układu.
Duże zainteresowanie środowiska naukowego oraz przemysłu związane z syntezą peptydów, spowodowało dynamiczny rozwój rynku syntezatorów. Automatyczne syntezatory są produkowane przez około 10 firm na świecie. Do najważniejszych producentów należą firmy CEM, AAPPTec, Activotec, Biotage, CEM, CSBio, Gyros Protein Technologies, Intavis Biotage. W ofercie znajdują się syntezatory dedykowane do prowadzenia procesów w skali laboratoryjnej (do kilku milimoli), a także te pozwalające na prowadzenie syntezy w skali przekraczającej 5 moli (CSBio, model CS936, rys. 3. A). W celu zapewnienia właściwego mieszania stosowane są mieszadła magnetyczne, mieszanie orbitalne oraz barbotaż, czyli przepuszczanie gazu obojętnego (azotu) przez mieszaninę reagentów. Standardowym rozwiązaniem jest synteza w jednym naczyniu reakcyjnym. Niemniej jednak oferowane są również syntezatory umożliwiające prowadzenie kilku(set) syntez jednocześnie. W celu otrzymywania bibliotek związków najwygodniej jest zastosować taki syntezator, który wykorzystuje odpowiednie płytki 96- i 384-dołkowe. Podejście to pozwala na otrzymywanie serii analogów związków w bardzo krótkim czasie. Co istotne, za przyspieszenie reakcji i zwiększenie wydajności odpowiedzialne jest podniesienie temperatury mieszaniny. Odbywać się to może poprzez umieszczenie naczynia reakcyjnego w polu promieniowania elektromagnetycznego z zakresu promieniowania mikrofalowego, z zakresu podczerwieni, a także poprzez tradycyjne ogrzewanie ścianek naczynia i następującej w konsekwencji konwekcji ciepła. Pojedyncze sprzęganie w przypadku syntezatorów mikrofalowych trwa około 4 minut (typowa synteza manualna – ok. 1-2 godziny). W syntezatorach firmy CEM (Liberty Blue, rys. 3, B) promieniowanie mikrofalowe stosowane jest zarówno podczas sprzęgania jak i deprotekcji ugrupowania Fmoc. Warunki te zostały określone mianem HE-SPPS7 (ang. High-efficiency Solid-phase peptide synthesis), a ich zastosowanie pozwalana na otrzymanie peptydu składającego się z kilkunastu reszt aminokwasowych w ciągu zaledwie godziny (więcej informacji: W. Kamysz, D. Grzywacz, Synteza mikrofalowa peptydów, Laborant, 9/2014).8

Rys. 3. Syntezatory peptydów: A) CSBio, model CS936;9 B) CEM, Liberty Blue;10 C) AAPPTec, Lab Mate.11
*na rysunku nie zachowano skali.
Syntezatory nie muszą być w pełni zautomatyzowane. Dostępne są także systemy pół-automatyczne (AAPPTec, Lab Mate, rys. 3. C), które co prawda wymagają większego zaangażowania w proces syntezy, lecz jednocześnie pozwalają na lepszą jego kontrolę.
Synteza w przepływie
Poza metodami syntezy opisanymi powyżej istnieje także możliwość prowadzenia jej w systemach przepływowych, w których żywica w sposób ciągły jest przemywana strumieniem reagentów i rozpuszczalników o zmiennym składzie zależnym od specyfiki danego etapu syntezy.12 Reagenty są przepompowywane przez kolumnę, w której umieszczona jest peptydylo-żywica. Wraz z przepływem pojawiają się istotne opory, a tym samym ciśnienie. W związku z tym stosowana żywica powinna charakteryzować się znaczną odpornością mechaniczną. Do tego typu syntezy stosuje się modyfikowaną żywicę jaką jest na przykład Tentagel, który na powierzchni usieciowanego polistyrenu posiada łańcuchy glikolu polietylenowego (PEG), zaś jego zawartość sięga 50-70% masy żywicy. Dodatkową korzyścią płynącą z tej modyfikacji jest zredukowanie zależności pęcznienia (objętości) żywicy od rodzaju rozpuszczalników, co jest szczególnie istotne w zestawieniu z ograniczoną przestrzenią wewnątrz kolumny. Także i w tym przypadku optymalizacja ma prowadzić do skrócenia czasu reakcji. W dużym uproszczeniu opiera się ona na zwiększeniu przepływu, podniesieniu temperatury układu i właściwym doborze reagentów. W efekcie tych działań opracowano systemy zdolne do przyłączenia kolejnych reszt aminokwasowych w czasie ok. 1,8 minuty (2014)13, a nawet 0,67 minuty (2017).14 Czas cyklu jest więc krótszy niż ten osiągany przez standardowe syntezatory automatyczne.
Innym z rozwijających się obszarów pokrewnych jest synteza w mikroukładach. Umożliwia ona otrzymywanie w sposób automatyczny niewielkich ilości związku, w relatywnie krótkim czasie i przy niewielkim zużyciu odczynników. Jednym z rozwiązań tego typu jest mikroreaktor zbudowany ze szkła borokrzemianowego, w którym ruch reagentów został osiągnięty za pomocą przyłożonego napięcia zewnętrznego (efekt elektroosmotyczny).15 Kolejną z możliwości jest prowadzenie reakcji w mikrochipie (tzw. Lab on chip). Układ ten składa się z systemu sterującego, pompy i chipu (mikroukładu), który zawiera liczne kanały wlotowe połączone z komorą reakcyjną oraz kanał wylotowy. Istotnym czynnikiem, który należy wziąć pod uwagę podczas projektowania mikro-chipu jest tworzywo, z którego ma być wykonany. Jest to o tyle istotne, że rozpuszczalniki stosowane w syntezie peptydów należą do „agresywnych” i znaczna część tworzyw pod ich wpływem ulega zniszczeniu. Jednym z rozwiązań jest wykorzystanie polimeru perfluoroalkoksylowego (PFA; kopolimer tetrafluoroetenu i eteru perfluoroalkilowinylowego).16
Podejmowane są także wysiłki by prowadzić równoległą syntezę peptydów w warunkach przepływowych na papierze modyfikowanym teflonem w celu otrzymywania bibliotek związków.17 Wydajność zachodzących procesów została zwiększona poprzez zastosowanie podwyższonej temperatury (promieniowanie podczerwone).18 Wspomaganie reakcji poprzez ogrzewanie może rozbudzać entuzjazm, jednakże należy mieć na uwadze to, że nie zawsze jest to droga prowadząca do sukcesu. Istotnym mankamentem jest zjawisko przedwczesnego odszczepienia peptydu od żywicy, co może w najlepszym przypadku obniżyć wydajność reakcji. Problem ten odnotowano w stosunku do żywicy 2-chlorotrytylowej (2-CTC), która z tego właśnie względu nie jest zalecana do prowadzenia syntez ze wspomaganiem mikrofalowym.19
Problem agregacji
Podczas syntezy często pojawia się problem niecałkowitego przyłączenia reszty aminokwasowej oraz niekompletnej deprotekcji. Może to być spowodowane agregacją łańcuchów peptydylo-żywicy, szczególnie w przypadku fragmentów bogatych w reszty aminokwasów hydrofobowych. W tworzeniu agregatów biorą udział wiązania wodorowe łańcucha peptydowego. Występowanie tego zjawiska skutkuje utrudnionym dostępem do miejsca reakcji, a w związku z tym obniżeniem jej wydajności. Jednym ze środków zaradczych jest zastosowanie podwyższonej temperatury, na przykład poprzez promieniowanie z zakresu mikrofalowego. Łańcuch peptydowy, ze względu na posiadany makrodipol oraz poszczególne dipole wiązań peptydowych, oddziałuje z oscylującym polem elektromagnetycznym. W efekcie ulega on rozprostowaniu, zaś grupy biorące udział w reakcji są eksponowane do środowiska, przez co niwelowany jest niekorzystny wpływ agregacji.20 Ponadto agregację można zminimalizować stosując dodatek rozpuszczalników, takich jak DMSO, NMP, THF, niejonowych surfaktantów (np. Triton X-100, Tween 20), soli chaotropowych (np. LiCl, MgBr2), a także poprzez obniżenie stopnia osadzenia żywicy, czy też sonikację mieszaniny reakcyjnej.21 Innym podejściem jest wykorzystanie odpowiednio modyfikowanych pochodnych aminokwasów. Jedną ze znanych modyfikacji są dipeptydowe pochodne bazujące na „pseudoprolinie”, a ściślej na strukturze 2,2-dimetyloksazolidyny i 2,2-dimetyltiazolidyny, które mogą być wytworzone przy udziale reszt seryny, treoniny oraz cysteiny (rys. 4).
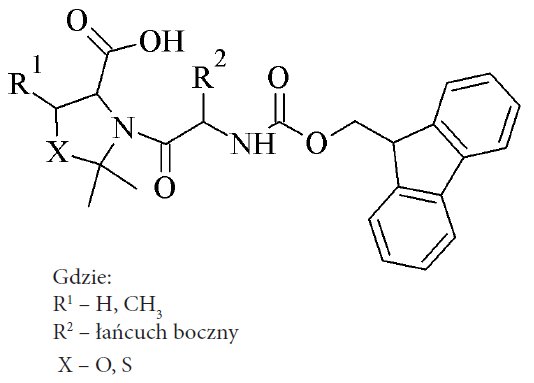
Rys. 4. Wzór pochodnej opierającej się na strukturze „pseudoproliny”.
Ryzyko agregacji łańcuchów jest obniżone ze względu na fakt występowania konfiguracji cis wiązania peptydowego w tejże pochodnej. Konfiguracja ta przerywając ciąg, który stanowią wiązania amidowe o konfiguracji trans, zaburza tworzenie się struktur wyższego rzędu. Ponadto pochodne zawierające ugrupowania 2,4-dimetoksybenzylu (Dmb), 2-hydroksy-4-metoksybenzylu (Hmb),22 i 2-hydroksy-4-metoksy-5-metylosulfinylobenzylu (Hmsb)23, które przyłączone są kowalencyjnie do atomu azotu chronionego jednocześnie ugrupowaniem Fmoc (Rys. 5) mogą skutecznie zapobiec tworzeniu się agregatów.
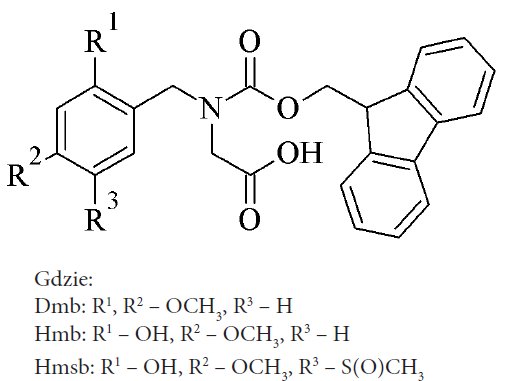
Rys. 5. Wzór strukturalny Fmoc-(Hmb/Dmb/Hmsb)Gly-OH.
Pochodne te nie zawierają donora wiązania wodorowego jakim jest ugrupowanie N-H (wiązanie peptydowe). W ten sposób uniemożliwione jest tworzenie się struktur β-kartki, odpowiedzialnych za agregację. Jednakże należy mieć na uwadze to, że zastosowanie pochodnej z ugrupowaniem Hmb jest obarczone ryzykiem tworzenia się laktonu, gdyż aktywowana grupa karboksylowa może reagować z grupą hydroksylową. Ponadto by odszczepić ugrupowanie Hmsb wymagane jest zastosowanie środowiska redukującego.23
Grupy ochronne na ratunek
Zastosowanie odpowiednich grup ochronnych w łańcuchach bocznych aminokwasów jest aspektem kluczowym dla powodzenia syntezy. Należy uwzględnić czy i w jaki sposób chcemy zabezpieczyć odpowiednie ugrupowania. Wybór ten może zaważyć na efektywności sprzęgania, deprotekcji, agregacji, a nawet ryzyku racemizacji. Szczególnie podatne na racemizację są pochodne cysteiny oraz histydyny. W celu ograniczenia tego niekorzystnego zjawiska należy wystrzegać się wysokich temperatur podczas reakcji sprzęgania, tj. przekraczających 50°C.24 Bardziej aktywnym podejściem do tego wyzwania jest stosowanie odpowiednich pochodnych chronionych ugrupowaniami obniżającymi stopień racemizacji wrażliwych reszt aminokwasowych. Przykładem takiej komercyjnie dostępnej pochodnej jest Fmoc-Cys(Thp)-OH (Rys. 6).
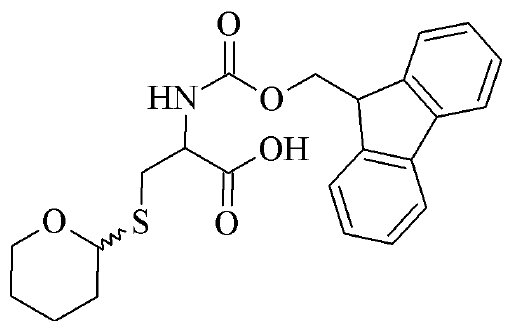
Rys. 6. Wzór strukturalny Fmoc-Cys(Thp)-OH.
Tetrahydropiranyl jest kwasolabilną grupą, która będąc przyłączoną kowalencyjnie do atomu siarki, skutecznie niweluje problem racemizacji.
H. Hibino i in. (2014)25 określili stopień racemizacji reszty cysteiny dla modelowego peptydu (Gly-Cys-Phe-NH2) stosując różne grupy ochronne i warunki syntezy. Część rezultatów została zamieszczona w tabeli 1, zaś na rys. 7 przedstawiono wzory strukturalne poszczególnych ugrupowań.
Tab. 1. Stopień racemizacji w odniesieniu do poszczególnych pochodnych cysteiny (grup ochronnych).
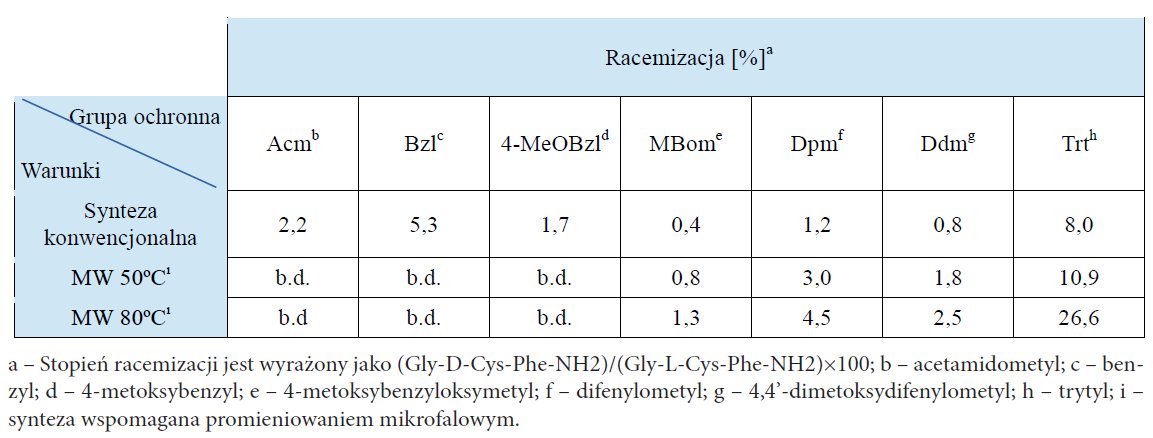
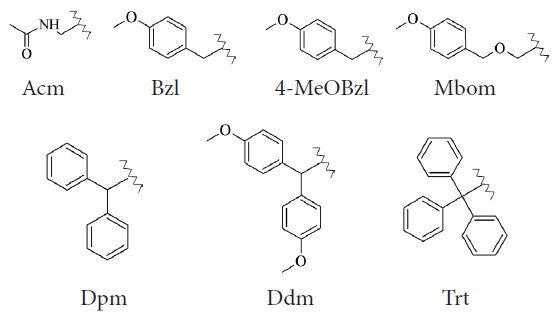
Rys. 7. Wzory strukturalne grup ochronnych łańcucha bocznego cysteiny.
Poza rodzajem zastosowanej pochodnej, istotne znaczenie mają także warunki syntezy oraz reagenty użyte podczas reakcji sprzęgania. Należy nadmienić, że korzystny efekt można uzyskać obniżając polarność środowiska, na przykład poprzez zmniejszenie udziału DMF na rzecz mniej polarnego DCM.26
Peptydy poza strukturą pierwszorzędową wykazują także znaczne zróżnicowanie pod względem przyjmowanych struktur wyższego rzędu. Znaczący udział w ustrukturyzowaniu mają wiązania dwusiarczkowe, które wytwarzane są pomiędzy dwoma resztami cysteiny. Peptydy mogą posiadać wiele wiązań tego typu. Na przykład α- i β-defensyny, będące peptydami o aktywności przeciwdrobnoustrojowej stanowiącymi element odporności nieswoistej, posiadają trzy wiązania disulfidowe. Sposób w jaki połączone są reszty cysteiny jest nie bez znaczenia. Właściwe wytworzenie wiązań w warunkach laboratoryjnych może jednak powodować trudności. Jak łatwo się domyślić, wraz ze wzrostem liczby wiązań disulfidowych zwiększa się złożoność syntezy. By otrzymać pożądany produkt z zadowalającą wydajnością zwykle niezbędne jest zastosowanie ortogonalnych grup ochronnych. Nie jest to jednak niezawodne rozwiązanie. Często stosowaną grupą ochronną jest tert-butyltiol (StBu), której usunięcie wymaga zastosowania środowiska o charakterze redukującym i zajmuje sporo czasu (do kilku godzin), co może nieść ze sobą ryzyko degradacji peptydu. Należy mieć na uwadze konieczność zachowania odpowiedniej kolejności podczas wytwarzania kolejnych wiązań dwusiarczkowych, które podczas deprotekcji grupy „StBu” mogłyby zostać zredukowane. Z tego względu wymusza ona pierwszeństwo, gdy jest stosowana wraz z innymi pochodnymi cysteiny. W celu zminimalizowania ryzyka wystąpienia reakcji ubocznych wprowadzono nowe grupy ochronne, których usunięcie zachodzi w krótszym czasie i w łagodniejszych warunkach. Jednym z nich jest ugrupowanie 2,4,6-trimetoksyfenylotiolowe (STmp), które można skutecznie usunąć stosując 5% ditiotreitol w DMF przez 5 minut.27
Piperydyna i kwestie zasadnicze
Etap deprotekcji jest przeprowadzany w celu uzyskania wolnych grup aminowych, które następnie mogą brać udział w reakcji sprzęgania. Zwykle do tego celu stosowany jest 20% roztwór piperydyny w DMF. Środowisko zasadowe tego roztworu, może wpływać niekorzystnie na bieg reakcji ze względu na racemizację oraz inne poważne reakcje uboczne. Przykładem jest powstawanie dehydroalaniny (Dha) z odpowiednio chronionej reszty cysteiny (szczególnie Acm). Pod wpływem piperydyny dochodzi do reakcji β-eliminacji. Możliwa jest także addycja piperydyny do powstałego wiązania podwójnego z wytworzeniem adduktu 3-(1-piperydynylo)alaniny.28 Kolejną reakcją uboczną, której przyczyną jest zasadowe środowisko roztworu piperydyny jest tworzenie się cyklicznego imidu z reszty kwasu asparaginowego. Jego powstawanie jest także wynikiem działania silnie kwaśnego środowiska, np. kwasu trifluorooctowego stosowanego w końcowym etapie syntezy. W efekcie może dojść do zmiany konfiguracji chiralnego atomu węgla, powstania β-peptydu, a także przyłączenia cząsteczki piperydyny. Szczególnie narażone na tego typu reakcje są peptydy zawierające fragment Asp-X, gdzie X jest resztą Gly, Asn, Gln, Asp, Arg, Ser, Thr, lub Ala.29 Ponadto podwyższona temperatura może istotnie sprzyjać zachodzeniu tej reakcji ubocznej. By ją zniwelować stosuje się odpowiednie pochodne dipeptydów do syntezy, np. Fmoc-Asp(OtBu)-(Dmb)Gly-OH i Asp(OtBu)-(Hmb)Gly-OH. W przypadku takich pochodnej atom azotu współtworzący wiązanie peptydowe jest kowalencyjnie związany z ugrupowaniem Dmb lub Hmb, co udaremnia ewentualny atak nukleofilowy na węgiel karbonylowy łańcucha bocznego Asp. Z drugiej strony stosowane są także zabezpieczenia samego łańcucha bocznego. Typową pochodną wykorzystywaną w syntezie jest Fmoc-L-Asp(OtBu)-OH. Wykazano, że grupy o zwiększonej zawadzie sterycznej obniżają ryzyko powstawania cyklicznego imidu.30 Można więc na tej podstawie dokonać swoistego uszeregowania grup ochronnych pod względem ryzyka zajścia wspomnianej reakcji ubocznej: OtBu > OMpe > OEpe > OBno. Pełne nazwy i wzory strukturalne przedstawiono na Rys. 8.
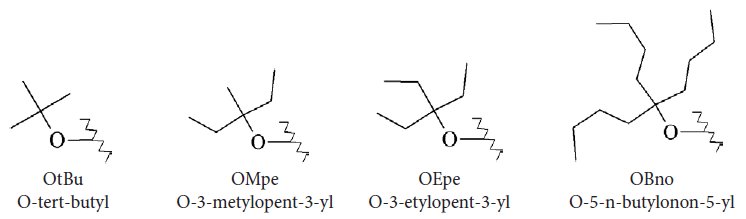
Rys. 8. Grupy ochronne stosowane w celu modyfikacji reszty kwasu asparaginowego.
Ponadto szanse na zajście reakcji ubocznej można skutecznie zmniejszyć stosując dodatek HOBt (1-hydroksybenzotriazol), HOAt (7-aza-1-hydroksybenzotriazol) lub Oxymy (2-cyjano2-(hydroksyimino)octan etylu) do roztworu piperydyny w DMF. Należy mieć na uwadze to, że związki wymienione powyżej posiadają charakter kwasowy. Wiąże się z tym możliwość przedwczesnego odszczepienia peptydu, gdy połączony jest on z linkerem żywicy za pomocą kwasolabilnego wiązania (żywica 2-CTC).31 Ponadto HOAt, HOBt, Oxyma należą do grupy związków ograniczających racemizację. Dlatego też są często stosowane podczas reakcji sprzęgania. Innymi związkami wykorzystywanymi do tego celu są m.in. Oxyma-B32 oraz K-Oxyma. Ten ostatni zalecany jest do stosowania podczas prowadzenia syntezy na żywicy 2-CTC. W tym przypadku, problem przedwczesnego odszczepienia został rozwiązany poprzez przeprowadzenie Oxymy w sól potasową (K-Oxyma), w wyniku czego zniwelowano charakter kwasowy tego reagenta przy zachowaniu aktywności przeciwracemizacyjnej. Dodatkowo zwiększeniu uległa stabilność temperaturowa oraz rozpuszczalność związku.33
Podsumowanie
Synteza peptydów na nośniku stałym rozwija się dynamicznie od kilkudziesięciu lat. Wraz z upływem czasu pokonywane są kolejne ograniczenia, zaś popularne rozwiązania są wciąż udoskonalane. Syntezatory peptydów umożliwiają otrzymywanie związków w coraz krótszym czasie i z wysoką wydajnością, przekraczającą możliwości syntezy manualnej/tradycyjnej. Z drugiej strony istnieje szereg związków, w powstawaniu których obecność człowieka jest niezbędna. Bardzo drogie pochodne aminokwasowe, tzw. trudne sekwencje w łańcuchu peptydowym czy problemy z oczyszczaniem finalnych produktów powodują, że kluczowym dla osiągnięcia sukcesu nadal są specjalistyczna wiedza i doświadczenie „peptydowca”.
Literatura:
(1) Schilbach, K.; Schopohl, J. Update on the Use of Oral Octreotide Therapy for Acromegaly. Expert Rev. Endocrinol. Metab. 2016, 1–7.
(2) Yeong, C.-H.; Cheng, M.; Ng, K.-H. Therapeutic Radionuclides in Nuclear Medicine: Current and Future Prospects. J. Zhejiang Univ. Sci. B 2014, 15 (10), 845–863.
(3) Galanis, A. S.; Albericio, F.; Grøtli, M. Solid-Phase Peptide Synthesis in Water Using Microwave-Assisted Heating. Org. Lett. 2009, 11 (20), 4488–4491.
(4) Gabriel, C. M.; Keener, M.; Gallou, F.; Lipshutz, B. H. Amide and Peptide Bond Formation in Water at Room Temperature. Org. Lett. 2015, 17 (16), 3968–3971.
(5) Http://peptideweb.com/index.php/peptide-Synthesis-Glassware-and-Reactors/solid-Phase-Peptide-Synthesis-Vessel-Side-Thread. Dostęp on-line 19-04-2017.
(6) Http://www.peptideweb.com/index.php/offer/shakers-and-Stirrers. Dostęp on-line 19-04-2017.
(7) Collins, J. M.; Porter, K. A.; Singh, S. K.; Vanier, G. S. High-Efficiency Solid Phase Peptide Synthesis ( HE -SPPS). Org. Lett. 2014, 16 (3), 940–943.
(8) Http://laborant.pl/index.php/synteza-Mikrofalowa-Peptydow.
(9) Http://www.csbio.com/wp-content/uploads/2013/09/2A.jpg. Dostęp on-line 19-04-2017.
(10) Http://cem.com/liberty-Blue/. Dostęp on-line 19-04-2017.
(11) Http://www.aapptec.com/lab-Mate-I-112.html. Dostęp on-line 19-04-2017.
(12) Lukas, T. J.; Prystowsky, M. B.; Erickson, B. W. Solid-Phase Peptide Synthesis under Continuous-Flow Conditions. Proc. Natl. Acad. Sci. U. S. A. 1981, 78 (5), 2791–2795.
(13) Simon, M. D.; Heider, P. L.; Adamo, A.; Vinogradov, A. A.; Mong, S. K.; Li, X.; Berger, T.; Policarpo, R. L.; Zhang, C.; Zou, Y.; Liao, X.; Spokoyny, A. M.; Jensen, K. F.; Pentelute, B. L. Rapid Flow-Based Peptide Synthesis. Chembiochem 2014, 15 (5), 713–720.
(14) Mijalis, A. J.; Thomas, D. A.; Simon, M. D.; Adamo, A.; Beaumont, R.; Jensen, K. F.; Pentelute, B. L. A Fully Automated Flow-Based Approach for Accelerated Peptide Synthesis. Nat. Chem. Biol. 2017, 13 (5), 464–466.
(15) Watts, P.; Wiles, C.; Haswell, S. J.; Pombo-Villar, E.; Styring, P. The Synthesis of Peptides Using Micro Reactors. In Microreaction Technology; Springer Berlin Heidelberg: Berlin, Heidelberg, 2001; pp 508–517.
(16) Zheng, H.; Wang, W.; Li, X.; Wang, Z.; Hood, L.; Lausted, C.; Hu, Z. An Automated Teflon Microfluidic Peptide Synthesizer. Lab Chip 2013, 13 (17), 3347.
(17) Deiss, F.; Matochko, W. L.; Govindasamy, N.; Lin, E. Y.; Derda, R. Flow-Through Synthesis on Teflon-Patterned Paper To Produce Peptide Arrays for Cell-Based Assays. Angew. Chemie Int. Ed. 2014, 53 (25), 6374–6377.
(18) Deiss, F.; Yang, Y.; Matochko, W. L.; Derda, R.; Mong, S. K.; Li, X.; Berger, T.; Policarpo, R. L.; Zhang, C.; Zou, Y.; Liao, X.; Spokoyny, A. M.; Jensen, K. F.; Pentelute, B. L.; Burton, D. R.; Wilson, I. A.; Cummings, R.; Bovin, N.; Wong, C.-H.; Paulson, J. C. Heat-Enhanced Peptide Synthesis on Teflon-Patterned Paper. Org. Biomol. Chem. 2016, 14 (22), 5148–5156.
(19) Echalier, C.; Al-Halifa, S.; Kreiter, A.; Enjalbal, C.; Sanchez, P.; Ronga, L.; Puget, K.; Verdié, P.; Amblard, M.; Martinez, J.; Subra, G. Heating and Microwave Assisted SPPS of C-Terminal Acid Peptides on Trityl Resin: The Truth behind the Yield. Amino Acids 2013, 45 (6), 1395–1403.
(20) Collins, J. M.; Leadbeater, N. E. Microwave Energy: A Versatile Tool for the Biosciences. Org. Biomol. Chem. 2007, 5 (8), 1141.
(21) Paradís-Bas, M.; Tulla-Puche, J.; Albericio, F. The Road to the Synthesis of "difficult Peptides". Chem. Soc. Rev. 2016, 45 (3), 631–654.
(22) Simmonds, R. G. Use of the Hmb Backbone-Protecting Group in the Synthesis of Difficult Sequences. Int. J. Pept. Protein Res. 1996, 47 (1–2), 36–41.
(23) Abdel-Aal, A.-B. M.; Papageorgiou, G.; Quibell, M.; Offer, J.; Mong, S. K.; Li, X.; Berger, T.; Policarpo, R. L.; Zhang, C.; Zou, Y.; Liao, X.; Spokoyny, A. M.; Jensen, K. F.; Pentelute, B. L. Automated Synthesis of Backbone Protected Peptides. Chem. Commun. 2014, 50 (61), 8316.
(24) Palasek, S. A.; Cox, Z. J.; Collins, J. M. Limiting Racemization and Aspartimide Formation in Microwave-Enhanced Fmoc Solid Phase Peptide Synthesis. J. Pept. Sci. 2007, 13 (3), 143–148.
(25) Hibino, H.; Miki, Y.; Nishiuchi, Y. Evaluation of Acid-Labile S-Protecting Groups to Prevent Cys Racemization in Fmoc Solid-Phase Peptide Synthesis. J. Pept. Sci. 2014, 20 (1), 30–35.
(26) Han, Y.; Albericio, F.; Barany, G. Occurrence and Minimization of Cysteine Racemization during Stepwise Solid-Phase Peptide Synthesis(1)(,)(2). J. Org. Chem. 1997, 62 (13), 4307–4312.
(27) Postma, T. M.; Giraud, M.; Albericio, F. Trimethoxyphenylthio as a Highly Labile Replacement for Tert -Butylthio Cysteine Protection in Fmoc Solid Phase Synthesis. Org. Lett. 2012, 14 (21), 5468–5471.
(28) Lukszo, J.; Patterson, D.; Albericio, F.; Kates, S. A. 3-(1-Piperidinyl)alanine Formation during the Preparation of C-Terminal Cysteine Peptides with the Fmoc/t-Bu Strategy. Lett. Pept. Sci. 1996, 3, 157–166.
(29) Bodanszky, M.; Kwei, J. Z. Side Reactions in Peptide Synthesis. VII. Sequence Dependence in the Formation of Aminosuccinyl Derivatives from Beta-Benzyl-Aspartyl Peptides. Int. J. Pept. Protein Res. 1978, 12 (2), 69–74.
(30) Behrendt, R.; Huber, S.; Martí, R.; White, P. New T -Butyl Based Aspartate Protecting Groups Preventing Aspartimide Formation in Fmoc SPPS. J. Pept. Sci. 2015, 21 (8), 680–687.
(31) Subirós-Funosas, R.; El-Faham, A.; Albericio, F. Use of Oxyma as pH Modulatory Agent to Be Used in the Prevention of Base-Driven Side Reactions and Its Effect on 2-Chlorotrityl Chloride Resin. Biopolymers 2012, 98 (2), 89–97.
(32) Jad, Y. E.; Khattab, S. N.; de la Torre, B. G.; Govender, T.; Kruger, H. G.; El-Faham, A.; Albericio, F. Oxyma-B, an Excellent Racemization Suppressor for Peptide Synthesis. Org. Biomol. Chem. 2014, 12 (42), 8379–8385.
(33) Cherkupally, P.; Acosta, G. A.; Nieto-Rodriguez, L.; Spengler, J.; Rodriguez, H.; Khattab, S. N.; El-Faham, A.; Shamis, M.; Luxembourg, Y.; Prohens, R.; Subiros-Funosas, R.; Albericio, F. K-Oxyma: A Strong Acylation-Promoting, 2-CTC Resin-Friendly Coupling Additive. European J. Org. Chem. 2013, 2013 (28), 6372–6378.
(1) Schilbach, K.; Schopohl, J. Update on the Use of Oral Octreotide Therapy for Acromegaly. Expert Rev. Endocrinol. Metab. 2016, 1–7.
(2) Yeong, C.-H.; Cheng, M.; Ng, K.-H. Therapeutic Radionuclides in Nuclear Medicine: Current and Future Prospects. J. Zhejiang Univ. Sci. B 2014, 15 (10), 845–863.
(3) Galanis, A. S.; Albericio, F.; Grøtli, M. Solid-Phase Peptide Synthesis in Water Using Microwave-Assisted Heating. Org. Lett. 2009, 11 (20), 4488–4491.
(4) Gabriel, C. M.; Keener, M.; Gallou, F.; Lipshutz, B. H. Amide and Peptide Bond Formation in Water at Room Temperature. Org. Lett. 2015, 17 (16), 3968–3971.
(5) Http://peptideweb.com/index.php/peptide-Synthesis-Glassware-and-Reactors/solid-Phase-Peptide-Synthesis-Vessel-Side-Thread. Dostęp on-line 19-04-2017.
(6) Http://www.peptideweb.com/index.php/offer/shakers-and-Stirrers. Dostęp on-line 19-04-2017.
(7) Collins, J. M.; Porter, K. A.; Singh, S. K.; Vanier, G. S. High-Efficiency Solid Phase Peptide Synthesis ( HE -SPPS). Org. Lett. 2014, 16 (3), 940–943.
(8) Http://laborant.pl/index.php/synteza-Mikrofalowa-Peptydow.
(9) Http://www.csbio.com/wp-content/uploads/2013/09/2A.jpg. Dostęp on-line 19-04-2017.
(10) Http://cem.com/liberty-Blue/. Dostęp on-line 19-04-2017.
(11) Http://www.aapptec.com/lab-Mate-I-112.html. Dostęp on-line 19-04-2017.
(12) Lukas, T. J.; Prystowsky, M. B.; Erickson, B. W. Solid-Phase Peptide Synthesis under Continuous-Flow Conditions. Proc. Natl. Acad. Sci. U. S. A. 1981, 78 (5), 2791–2795.
(13) Simon, M. D.; Heider, P. L.; Adamo, A.; Vinogradov, A. A.; Mong, S. K.; Li, X.; Berger, T.; Policarpo, R. L.; Zhang, C.; Zou, Y.; Liao, X.; Spokoyny, A. M.; Jensen, K. F.; Pentelute, B. L. Rapid Flow-Based Peptide Synthesis. Chembiochem 2014, 15 (5), 713–720.
(14) Mijalis, A. J.; Thomas, D. A.; Simon, M. D.; Adamo, A.; Beaumont, R.; Jensen, K. F.; Pentelute, B. L. A Fully Automated Flow-Based Approach for Accelerated Peptide Synthesis. Nat. Chem. Biol. 2017, 13 (5), 464–466.
(15) Watts, P.; Wiles, C.; Haswell, S. J.; Pombo-Villar, E.; Styring, P. The Synthesis of Peptides Using Micro Reactors. In Microreaction Technology; Springer Berlin Heidelberg: Berlin, Heidelberg, 2001; pp 508–517.
(16) Zheng, H.; Wang, W.; Li, X.; Wang, Z.; Hood, L.; Lausted, C.; Hu, Z. An Automated Teflon Microfluidic Peptide Synthesizer. Lab Chip 2013, 13 (17), 3347.
(17) Deiss, F.; Matochko, W. L.; Govindasamy, N.; Lin, E. Y.; Derda, R. Flow-Through Synthesis on Teflon-Patterned Paper To Produce Peptide Arrays for Cell-Based Assays. Angew. Chemie Int. Ed. 2014, 53 (25), 6374–6377.
(18) Deiss, F.; Yang, Y.; Matochko, W. L.; Derda, R.; Mong, S. K.; Li, X.; Berger, T.; Policarpo, R. L.; Zhang, C.; Zou, Y.; Liao, X.; Spokoyny, A. M.; Jensen, K. F.; Pentelute, B. L.; Burton, D. R.; Wilson, I. A.; Cummings, R.; Bovin, N.; Wong, C.-H.; Paulson, J. C. Heat-Enhanced Peptide Synthesis on Teflon-Patterned Paper. Org. Biomol. Chem. 2016, 14 (22), 5148–5156.
(19) Echalier, C.; Al-Halifa, S.; Kreiter, A.; Enjalbal, C.; Sanchez, P.; Ronga, L.; Puget, K.; Verdié, P.; Amblard, M.; Martinez, J.; Subra, G. Heating and Microwave Assisted SPPS of C-Terminal Acid Peptides on Trityl Resin: The Truth behind the Yield. Amino Acids 2013, 45 (6), 1395–1403.
(20) Collins, J. M.; Leadbeater, N. E. Microwave Energy: A Versatile Tool for the Biosciences. Org. Biomol. Chem. 2007, 5 (8), 1141.
(21) Paradís-Bas, M.; Tulla-Puche, J.; Albericio, F. The Road to the Synthesis of "difficult Peptides". Chem. Soc. Rev. 2016, 45 (3), 631–654.
(22) Simmonds, R. G. Use of the Hmb Backbone-Protecting Group in the Synthesis of Difficult Sequences. Int. J. Pept. Protein Res. 1996, 47 (1–2), 36–41.
(23) Abdel-Aal, A.-B. M.; Papageorgiou, G.; Quibell, M.; Offer, J.; Mong, S. K.; Li, X.; Berger, T.; Policarpo, R. L.; Zhang, C.; Zou, Y.; Liao, X.; Spokoyny, A. M.; Jensen, K. F.; Pentelute, B. L. Automated Synthesis of Backbone Protected Peptides. Chem. Commun. 2014, 50 (61), 8316.
(24) Palasek, S. A.; Cox, Z. J.; Collins, J. M. Limiting Racemization and Aspartimide Formation in Microwave-Enhanced Fmoc Solid Phase Peptide Synthesis. J. Pept. Sci. 2007, 13 (3), 143–148.
(25) Hibino, H.; Miki, Y.; Nishiuchi, Y. Evaluation of Acid-Labile S-Protecting Groups to Prevent Cys Racemization in Fmoc Solid-Phase Peptide Synthesis. J. Pept. Sci. 2014, 20 (1), 30–35.
(26) Han, Y.; Albericio, F.; Barany, G. Occurrence and Minimization of Cysteine Racemization during Stepwise Solid-Phase Peptide Synthesis(1)(,)(2). J. Org. Chem. 1997, 62 (13), 4307–4312.
(27) Postma, T. M.; Giraud, M.; Albericio, F. Trimethoxyphenylthio as a Highly Labile Replacement for Tert -Butylthio Cysteine Protection in Fmoc Solid Phase Synthesis. Org. Lett. 2012, 14 (21), 5468–5471.
(28) Lukszo, J.; Patterson, D.; Albericio, F.; Kates, S. A. 3-(1-Piperidinyl)alanine Formation during the Preparation of C-Terminal Cysteine Peptides with the Fmoc/t-Bu Strategy. Lett. Pept. Sci. 1996, 3, 157–166.
(29) Bodanszky, M.; Kwei, J. Z. Side Reactions in Peptide Synthesis. VII. Sequence Dependence in the Formation of Aminosuccinyl Derivatives from Beta-Benzyl-Aspartyl Peptides. Int. J. Pept. Protein Res. 1978, 12 (2), 69–74.
(30) Behrendt, R.; Huber, S.; Martí, R.; White, P. New T -Butyl Based Aspartate Protecting Groups Preventing Aspartimide Formation in Fmoc SPPS. J. Pept. Sci. 2015, 21 (8), 680–687.
(31) Subirós-Funosas, R.; El-Faham, A.; Albericio, F. Use of Oxyma as pH Modulatory Agent to Be Used in the Prevention of Base-Driven Side Reactions and Its Effect on 2-Chlorotrityl Chloride Resin. Biopolymers 2012, 98 (2), 89–97.
(32) Jad, Y. E.; Khattab, S. N.; de la Torre, B. G.; Govender, T.; Kruger, H. G.; El-Faham, A.; Albericio, F. Oxyma-B, an Excellent Racemization Suppressor for Peptide Synthesis. Org. Biomol. Chem. 2014, 12 (42), 8379–8385.
(33) Cherkupally, P.; Acosta, G. A.; Nieto-Rodriguez, L.; Spengler, J.; Rodriguez, H.; Khattab, S. N.; El-Faham, A.; Shamis, M.; Luxembourg, Y.; Prohens, R.; Subiros-Funosas, R.; Albericio, F. K-Oxyma: A Strong Acylation-Promoting, 2-CTC Resin-Friendly Coupling Additive. European J. Org. Chem. 2013, 2013 (28), 6372–6378.


