


Jacek Achrem-Achremowicz, Wyatt Technology Europe
Stepan Podzimek, Synpo, Pardubice, Republika Czeska
strony wersji drukowanej: 24-29
strony wersji drukowanej: 24-29
UHPLC - wprowadzenie
Techniki chromatograficzne są jednymi z najczęściej stosowanych metod instrumentalnych w laboratoriach. Ich rozwój warunkuje uzyskiwanie coraz lepszego rozdzielania próbek, z coraz większą dokładnością i w coraz krótszym czasie. Dynamika zmian w porównaniu np. z technologiami IT nie jest aż taka szybka, jednak pojawiają się tutaj znaczące przełomy. Jednym z ostatnich było zastosowanie bardzo małego uziarnienia złoża kolumn chromatograficznych. Ta pozornie niewielka modyfikacja wpłynęła bardzo istotnie na zwiększenie rozdzielczości i skrócenie czasu analiz; o rząd wielkości - do kilku minut. Średnica ziaren złoża w klasycznych kolumnach do wysokosprawnej chromatografii cieczowej (HPLC) wynosi ok. 5 μm. Zmniejszenie tej wartości poniżej 3 μm a obecnie nawet poniżej 2 μm wymagało zastosowania pomp i elementów systemu chromatograficznego zdolnych wytwarzać i pracować przy znacznie wyższych ciśnieniach. Ponieważ przy tak małym ziarnie wypełniającym kolumnę opór przepływu jest bardzo duży, wymagane ciśnienia są ponad 400 bar, częściej rzędu 600 a niekiedy 1000 bar. Skrót tego rodzaju chromatografii UHPLC tłumaczy się dwojako – ultrasprawna chromatografia cieczowa (Ultra High Performance Liquid Chromatography) lub ultra-wysokociśnieniowa chromatografia żelowa (Ultra High Pressure LC). Druga z tych nazw uwypukla czynnik ,,ciśnieniowy” w tej technice. Jedną z jej kluczowych korzyści tej nowej techniki chromatograficznej jest znaczące skrócenie czasu analiz. Wynika to bezpośrednio ze zmniejszenia czasów/objętości retencjianalizowanych próbek oraz czasu potrzebnego na stabilizację układu. Zastosowanie odpowiedniej długości/ilości kolumn pozwla na wzrost rozdzielczości, a co za tym idzie uzyskiwanie nowych informacji o składnikach próbki. Można w ten sposób np. potwierdzić wysoką czystość produktu bądź wykazać obecność innych substancji, które w klasycznej chromatografii cieczowej ,,ukrywałyby” się pod pikiem w formie np. słabo zaznaczonego rozmycia.
W chromatografii UHPLC, po rozdzieleniu na kolumnie, wymagane jest zastosowanie odpowiednich modułów detekcyjnych w celu identyfikacji i oznaczania wymywanych składników. Najczęściej stosowane są detektory UV-VIS oraz refraktometryczne, które dają wyśmienite wyniki w analizach związków niskocząsteczkowych dające się łatwo potwierdzić jakościowo i ilościowo zastosowaniem wzorców. Sprzężenie systemu ze spektrometrią mas daje szerszy zakres możliwości analitycznych, ale wymaga większego doświadczenia i wiedzy badacza.
Ultrasprawna chromatografia żelowa (UHP-SEC)
Zastosowanie specjalnych kolumn do chromatografii żelowej SEC/GPC (Size Exclusion Chromatography/Gel Permeation Chromatography) w systemie UHPLC pozwala na przeprowadzanie analiz związków makrocząsteczkowych, takich jak np. białka lub polimery syntetyczne. W przypadku tych klas związków od bezpośredniej identyfikacji (nie zawsze możliwej – np. brak standardów) bardziej interesujące staje się wyznaczenie ich mas cząsteczkowych lub dystrybucji/rozkładu mas cząsteczkowych w obrębie piku. W tym celu konieczna jest kalibracja kolumny serią standardów mas cząsteczkowych, aby wyznaczyć zależność: masa cząsteczkowa—czas/objętość retencji.
W chromatografii żelowej kolejność wymywania związana jest z zatrzymywaniem molekuł w porach złoża kolumny. Cząsteczki mniejsze od średnicy tych porów są w stanie wnikać do wnętrza ziaren natomiast większe są z nich całkowicie ,,wykluczane” i w trakcie wymywania przemieszczają się pomiędzy ziarnami złoża, w stronę wylotu kolumny. Ponieważ wszystkie cząsteczki jednocześnie podlegają dyfuzji, małe, które dostały się do wnętrza porów, po jakimś czasie wydostają się na zewnątrz i są transportowane przez fazę ruchomą w kierunku wylotu kolumny. Czas przebywania cząsteczek wewnątrz porów jest proporcjonalny do ich wielkości a ściślej objętości hydrodynamicznej (Vh) – im są mniejsze, tym dłużej są zatrzymywane wewnątrz porów i tym później wymywane.
Kalibracja w SEC i UHP-SEC jest podobna. Nastrzyknięcie serii wzorców o dobrze zdefiniowanych masach cząsteczkowych wyznaczonych innymi technikami1, pozwala na tzw. standardową kalibrację kolumny. Na podstawie porównania czasów retencji wymywanych składników i wzorców można oszacować masę cząsteczkową analitu. Uzyskane w ten sposób masy cząsteczkowe są tylko pewnym przybliżeniem. Związane jest to z przyjętymi założeniami: 1) masa cząsteczkowa ma określoną ścisłą proporcjonalność do objętości hydrodynamicznej, warunkującej kolejność wymywania; 2) w trakcie wymywania warunki ,,wykluczania” SEC są idealne, tzn. nie zachodzą oddziaływania składników próbki ze złożem kolumny inne niż objętościowe; 3) cząsteczki posiadające rozgałęzienia zachowują się tak jakby ich nie miały i ich wymywanie nie jest opóźniane przez zakotwiczanie luźnych gałęzi w porach złoża i między ziarnami.
W przypadku 1) stwierdzenie wydaje się całkiem prawidłowe. Trzeba jednak uwzględnić szereg wyjątków, gdzie zastosowano wzorzec o określonej geometrii i masie, a analizowane, nieznane związki, mają inny kształt. Powoduje to, że wyznaczone masy cząsteczkowe wypadają przed lub za standardem(ami). W tym przypadku to czy błąd wyniesie kilkadziesiąt czy kilkaset procent zależy od ,,trafności” dobrania wzorca. Przykład takiej sytuacji pokazano na Rysunku 2.
W większości przypadków dla analityka interesujące są dobrze zdefiniowane warunki chromatografowania. W HPLC potrzebne jest uzyskanie odpowiedniego, kontrolowanego oddziaływania (entalpowego) składników próbki ze złożem, natomiast w SEC wyeliminowanie tego pierwszego i uzyskanie rozdzielenia tylko ze względu na różnice wielkości wymywanych składników. W praktyce zaistnienie takich idealnych warunków nie jest możliwe i swój udział w rozdzieleniu mają obydwa wymienione czynniki (entalpowy i steryczny). Im bardziej różnią się od siebie wzorzec i badany składnik próbki tym założenie 2 jest mniej słuszne, gdyż występuje większa różnica w oddziaływaniach ze złożem kolumny, powodująca rosnące rozbieżności czasu retencji, a co za tym idzie różnice oszacowanych mas cząsteczkowych.
Założenie 3) dotyczy cząstek, których fragmenty ,,nie powinny” zaczepiać się w porach złoża, co jest niekiedy tak trudne w realizacji, że wymaga zastosowania innej techniki frakcjonowania niż chromatografia żelowa, jak np. ultrawirowanie (AU) lub frakcjonowanie w polu sił (FFF).
Pomimo ww. ograniczeń chromatografia żelowa jest bardzo popularna w badanich makromolekuł. Zastosowanie jako detektora mas cząsteczkowych w SEC fotometru MALS pozwala uzyskiwać wartości mas cząsteczkowych w sposób absolutny.

Rysunek 1. Pierwszy fotometr wielokątowego rozpraszania światła laserowego (MALS) – μDAWN firmy Wyatt Technology; zakres wyznaczanych mas cząsteczkowych od: 200 Da do 10 MDa (g/mol) dla białek i 200 Da-1 MDa dla polimerów liniowych; promień RMS (rg) 10-50 nm; opcjonalnie pomiar promieni hydrodynamicznych (rh) za pomocą DLS w trybie chromatograficznym – od 0,5-30 nm.
Korzyści sprzężenia UHP-SEC z detekcją absolutnych mas cząsteczkowych za pomocą MALS
Fotometr MALS mierzy całkowite natężenie światła rozproszonego przez roztwór makromolekuł pod kilkoma kątami jednocześnie. Instrument ten jest łączony kapilarą wychodzącą z systemu chromatograficznego, a pomiar rozpraszania światła dokonywany jest przez fotodiody umieszczone wokół celi przepływowej. Upraszczając rozważania teoretyczne, można powiedzieć, że intensywność tego rozproszenia jest wprost proporcjonalna do iloczynu masy cząsteczkowej i stężenia danej substancji. Zatem znając stężenie substancji w każdym punkcie chromatogramu i jednocześnie mierząc intensywność rozproszenia światła fotometrem MALS można uzyskać wartości mas cząsteczkowych wszystkich wymywanych składników próbki i to bez odnoszenia się do standardów mas cząsteczkowych (!). Uzyskanie rzeczywistych wartości stężeń jest możliwe z zastosowaniem np. różnicowego detektora refraktometrycznego (dRI), ale przy znanych wartościach współczynnika dn/dc, który jest na ogół znaną, stałą wartością, charakterystyczną dla danego związku (wartości dn/dc można również wyliczyć teoretycznie lub wyznaczyć w odrębnym eksperymencie z dRI).
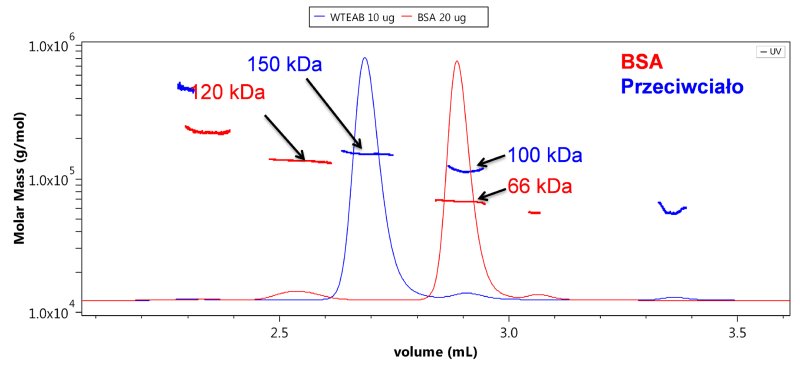
Rysunek 2. Klasyczny problem przy wyznaczaniu mas cząsteczkowych – kalibracja standardami daje błędne wyniki (o ponad 30% mniejsze), tymczasem detekcja MALS pozwala na wyznaczenie absolutnych, tj. niezależnych od standardów, mas cząsteczkowych. Przeciwciało (120 kDa; promień Rh 5,5 nm) wymywane jest wcześniej niż cięższy, ale mniejszy od niego dimer BSA (150 kDa; Rh 4,2 nm). Na chromatogramie pokazano masę cząsteczkową w funkcji objętości elucji oraz sygnał różnicowego detektora refraktometrycznego. Sygnały odpowiednio z μDAWN (MALS) i Optilab UT-rEX (dRI), obydwa Wyatt Technology, dedykowane do UHPLC.
Fundamentalną korzyścią połączenia fotometru MALS z systemem UHPLC-SEC (lub również z SEC) jest eliminacja błędnych założeń kalibracji standardowej i uzyskiwanie absolutnych mas cząsteczkowych dla każdego wymywanego związku. Daje to analitykowi bardzo potężne narzędzie w badaniach związków makrocząsteczkowych. Łączy ono zalety uzyskiwania rzeczywistych mas cząsteczkowych i ich rozkładu w obrębie polidyspersyjnych pików dzięki MALS z korzyściami systemu frakcjonującego UHPLC o bardzo wysokiej rozdzielczości i/lub przepustowości. Krótki czas analiz ułatwia np. niemal natychmiastową kontrolę procesów. Natomiast dodatkowe zwiększenie rozdzielczości UHPLC, które można uzyskać kosztem wydłużenia czasu rozdzielania (np. stosując 2 kolumny szeregowo), pozwala na bardzo szczegółową analizę chromatograficzną o niezwykłej czułości. Przykład uzyskania nowych informacji o fragmentacji znanego białka pokazano na Rysunku 3.
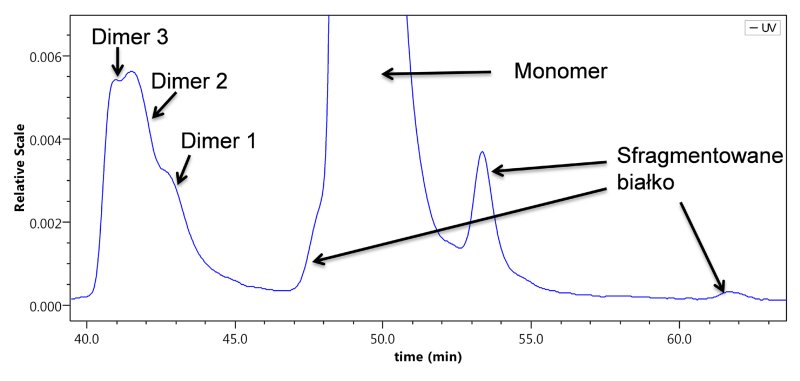
Rysunek 3. Nowe fakty dotyczące znanego białka: analiza UHP-SEC-MALS standardu białkowego BSA. Zastosowano 2 kolumny Waters Acquity SEC BEH 200; Vr =0,1 ml/min. Poprawne wyznaczenie mas cząsteczkowych pików fragmentacji i oligomerów możliwe jest tylko za pomocą detekcji techniką absolutną – tutaj MALS.
Włączenie dwóch detektorów stężeniowych do systemu chromatograficznego z fotometrem MALS – np. dRI oraz UV-VIS – daje możliwość badania mas cząsteczkowych kopolimerów typu (A)x(B)y.
Związkami takimi mogą być białka farmaceutyczne, np. glikozylowane lub PEG-ylowane. Dzięki takiej detekcji uzyskuje się precyzyjne wartości mas cząsteczkowych: białka (polimer A), części polisacharydowej (polimer B) oraz całej cząsteczki AB (Rysunek 4).
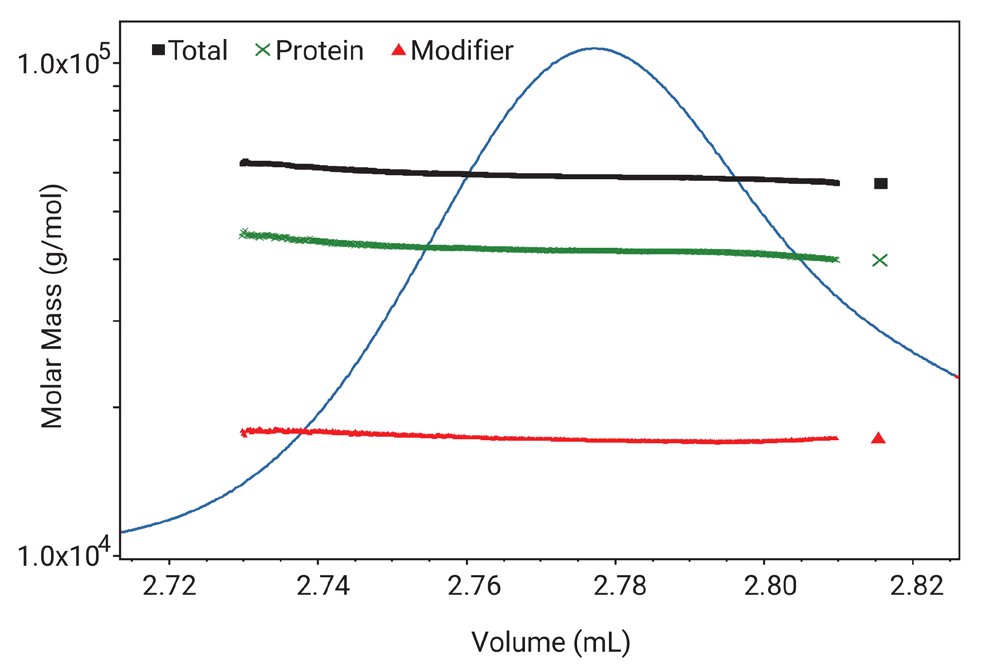
Rysunek 4. Połączenie detekcji: MALS, dRI i UV może być stosowane do analizy składu koniugatów białko-polisacharyd, białek membranowych w otoczce detergentu i niektórych kopolimerów. Na wykresie pokazano analizę glikoprotein za pomocą fotometru MALS: μDAWN (Wyatt Technology), refraktometru Optilab UT-rEX (Wyatt Technology) sprzężonych z systemem UHPLC Acquity (Waters) z kolumną BEH-200. Masa cząsteczkowa w funkcji objętości elucji jest stała, co świadczy o jednorodności składu glikozylowanego białka; od dołu są Mw części polisacharydowej (czerwony), białka (zielony) i całego kompleksu (czarny).
Tego rodzaju badania są niezwykle istotne min. dla niektórych działów badawczych firm farmaceutycznych, zajmujących się białkami medycznymi.
Fotometr MALS dedykowany do UHPLC
Fotometr MALS dedykowany do UHPLC
Aby rozdzielczość wąskich pików uzyskanych dzięki UHPLC została zachowana, poszerzenie pasm (dyspersja pików) w kolejnych sprzężonych za kolumną detektorach musi być jak najmniejsze. Dotyczy to również fotometru MALS. W połowie 2015 r. na rynku pojawił się pierwszy fotometr MALS μDAWN firmy Wyatt Technology (rys. 1), specjalnie zaprojektowany do podłączenia do najnowszych systemów chromatografii UHPLC. Zastosowano w nim celę przepływową o objętości poniżej 10 μl i cienkie kapilary, w bardzo małym stopniu wpływające na rozmywanie pasm. Oczywiście w detektorze tego typu, do uzyskiwania dobrych wyników nie jest wystarczające samo zmniejszenie celi przepływowej, ale konieczna jest także optymalizacja wielu parametrów technicznych. Wśród nich kluczowe znaczenie ma stosunek sygnału do szumu (S/N), który zależy od geometrii i jakości optoelektroniki, kształtu oraz materiału celi przepływowej i in. W nowym detektorze Wyatt zastosował swoje klasyczne, wyrafinowane rozwiązania techniczne, np. pomiar ciągły natężenia światła padającego na celę pomiarową (LM – Laser Monitor) i światła przez nią przechodzącego (FM – Forward Monitor). Te dodatkowe pomiary pozwalają na wyeliminowanie: efektów związanych z częściowym zabrudzeniem celki lub absorpcją światła przez składniki próbki, jak i fluktuacji natężenia światła padającego (powodowane np. przez zmiany napięcia w sieci, zużycie lasera). Zakres mierzonych absolutnie mas cząsteczkowych jest bardzo szeroki i wynosi 200 Da-10 MDa dla białek i 200 Da - 1 MDa dla polimerów liniowych. Dla cząsteczek o wielkościach ponad 10 nm można za pomocą fotometru MALS wyznaczać również tzw. promień bezwładności Rg (in. średnią kwadratową promienia - RMS). W uDAWN zakres pomiarowy Rg wynosi 10 do 50 nm (przykład na Rysunku 6).
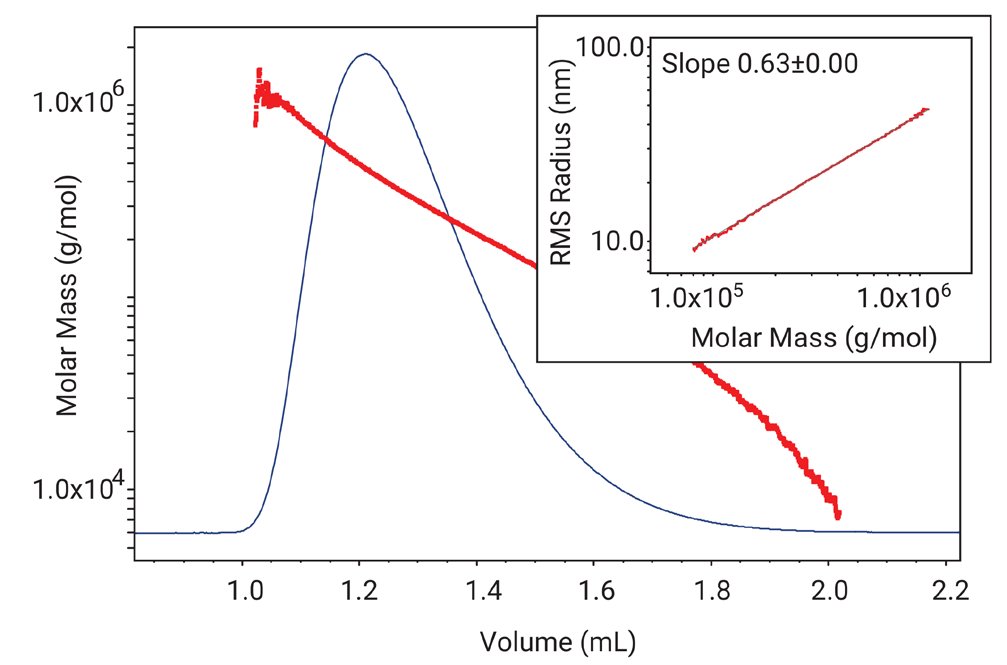
Rysunek 5. Analiza standardu polimerowego NIST 706a (polistyren) za pomocą UHPLC: Waters Acquity z kolumnami APC XT 900 i XT 450 oraz detekcją μDAWN (MALS) i UT-rEX (RI) – obydwa Wyatt Technology. Widoczna heterogenność próbki z masą cząsteczkową malejącą w całym zakresie pomiarowym. Na wstawce pokazano wykres konformacyjny – zależność promienia RMS względem masy cząsteczkowej. Nachylenie wykresu RMS=f(Mw) (tanΘ=0,63 na wstawce) świadczy, że polimer to liniowy polistyren o konformacji kłębka statystycznego.
Ponadto do instrumentu można opcjonalnie przyłączyć moduł QELS, do pomiaru dynamicznego rozpraszania światła (DLS), pozwalający na zmierzenie promieni hydrodynamicznych (Rh) od 0,5 do 30 nm. Pomiar DLS jest dokonywany jednocześnie z MALS w tej samej celi pomiarowej (Rysunek 6). Dodatkowe dane z DLS są często bardzo cenną informacją i umożliwiają np. zbadanie wielkości powstających agregatów lub tworzenie wykresów konformacyjnych, które mówią o kształtach cząsteczek i ich upakowaniu.
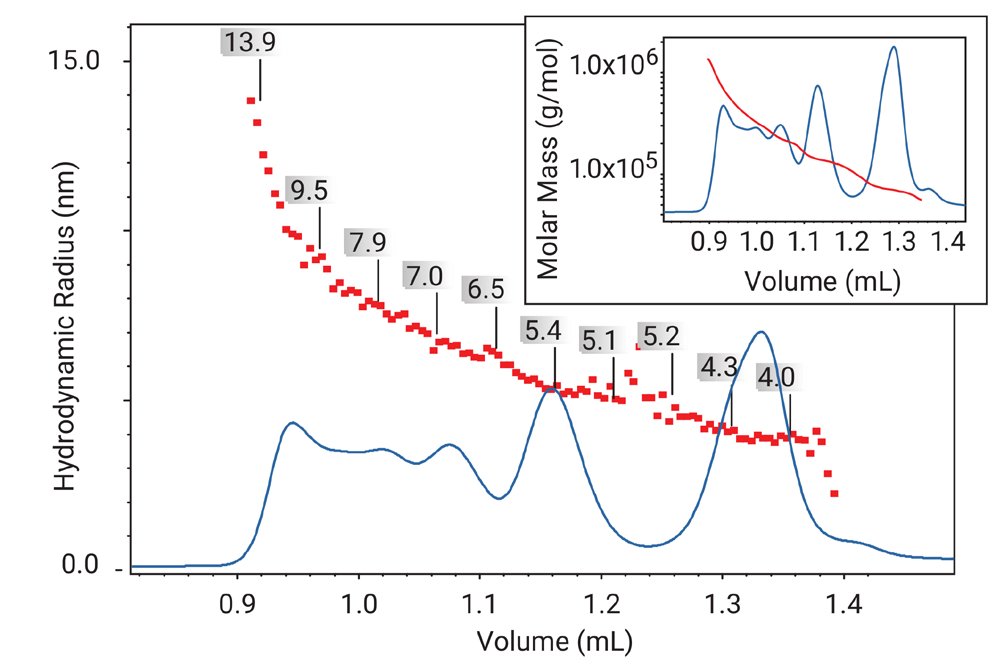
Rysunek 6. Wykres dystrybucji promieni hydrodynamicznych (rh) oraz na wstawce mas cząsteczkowych w funkcji objętości elucji (Vel) dla próbki białka BSA poddanego silnemu stresowi (dializa z 10 M mocznika do buforu fosforanowego). Dimer, trimer i tetramer zostały rozdzielone i zidentyfikowane na podstawie ich Mw.
Podsumowanie
Rozwojowi chromatograficznych technik separacyjnych towarzyszy udoskonalanie istniejących oraz powstawanie nowych detektorów, które są niezbędne do uzyskiwania poprawnych informacji o rozdzielonych składnikach. Jednym z takich instrumentów jest fotometr MALS dedykowany do ultrasprawnej chromatografii żelowej (UHP-SEC), pozwalający na wyznaczanie rzeczywistych mas cząsteczkowych w sposób absolutny, służący również do pomiaru wielkości cząsteczek w trybie chromatograficznym oraz innych badań opartych na MALS jak analizy: konformacyjne, koniugatów białkowych, białek błonowych. Aby lepiej zapoznać się z teoretycznymi i praktycznymi zagadnieniami MALS oraz sprzężenia tej techniki z systemami frakcjonującymi warto skorzystać z książki S.Podzimka: Light Scattering, Size Exclusion Chromatography and Asymmetric Flow Field Flow Fractionation: Powerful Tools for the Characterization of Polymers, Proteins and Nanoparticles (Wiley, 2011).
1Podawane przez producentów masy cząsteczkowe standardów są zawsze obarczone pewnym błędem, który się dalej propaguje.


