


Krzysztof Brzozowski
Uniwersytet Gdański, Wydział Chemii
Kilka słów wstępu
Budowa cząsteczki oraz zrozumienie zależności aktywność-struktura to jedno z kluczowych zadań dzisiejszej biochemii strukturalnej. Tytułowe peptydy i białka należą do grupy najczęściej badanych biopolimerów. Dzieje się tak ze względu na ich udział w wielu przemianach biochemicznych zachodzących w żywych organizmach. Główne funkcje białek pokazuje poniższa klasyfikacja:
- strukturalna – białka strukturalne są odpowiedzialne za kształt komórek i tkanek. Przykładem mogą być kolagen lub histony, które odpowiedzialne są za organizację DNA w chromatynie;
- transportowa – przykładem takiego białka jest hemoglobina odpowiedzialna za transport tlenu i dwutlenku węgla pomiędzy płucami i tkankami;
- ochronna – układ odpornościowy chroni ciało przed patogenami i obcymi substancjami. Ważnym jego składnikiem jest immunoglobulina G;
- regulatorowa – w kaskadzie biochemicznej białka pełnią funkcje sygnałowe (hormony) i receptorowe. Np. połączenie somatotropiny z jej receptorem powoduje aktywację domeny cytoplazmatycznej i prowadzi do przesłania sygnału do wnętrza komórki;
- katalityczna – enzymy stanowią największą grupę białek (ponad 2000 znanych biomolekuł).
- mobilna – oddziaływania pomiędzy aktyną i miozyną są odpowiedzialne za kontrakcję mięśni i ruchy komórkowe;
- magazynowa – rośliny zawierają specjalne białka, które są ważne dla człowieka z żywieniowego punktu widzenia. Przykładem może być kazeina lub ferytyna.
Dlaczego warto użyć tego narzędzia
Konformacja łańcucha polipeptydowego może być całkowicie zdefiniowana poprzez określenie orientacji dwóch wiązań peptydowych przyłączonych bezpośrednio do tego samego atomu Cα – rysunek 1.
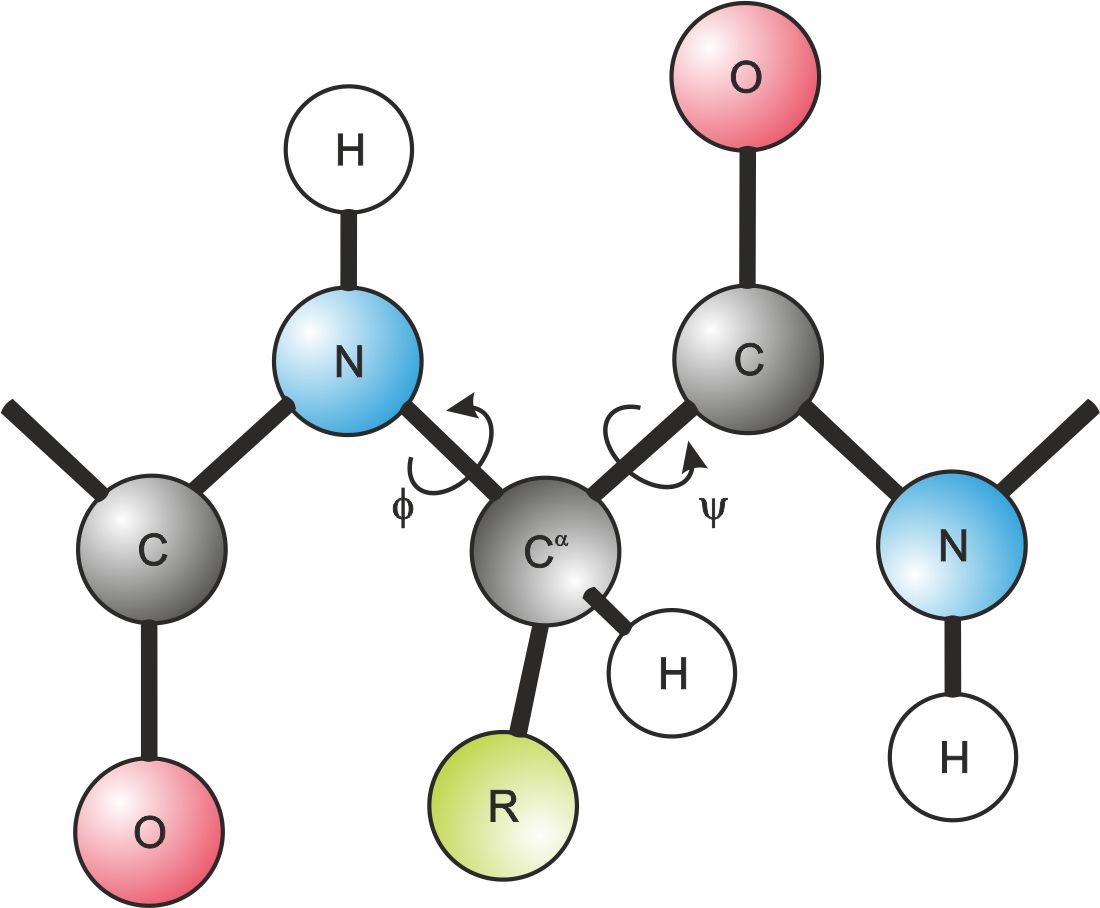
Rysunek 1. Kąty określające orientację dwóch wiązań peptydowych przyłączonych do tego samego atomu Cα.
Rysunek 1. Kąty określające orientację dwóch wiązań peptydowych przyłączonych do tego samego atomu Cα.
Badając różne kombinacje kątów dwuściennych ϕ i ψ Ramakrishan i Ramachandran zaproponowali wykres wzajemnych zależności tych dwóch wartości [1]. Wyróżnili oni obszary odpowiadające poszczególnym elementom struktury drugorzędowej – rysunek 2.
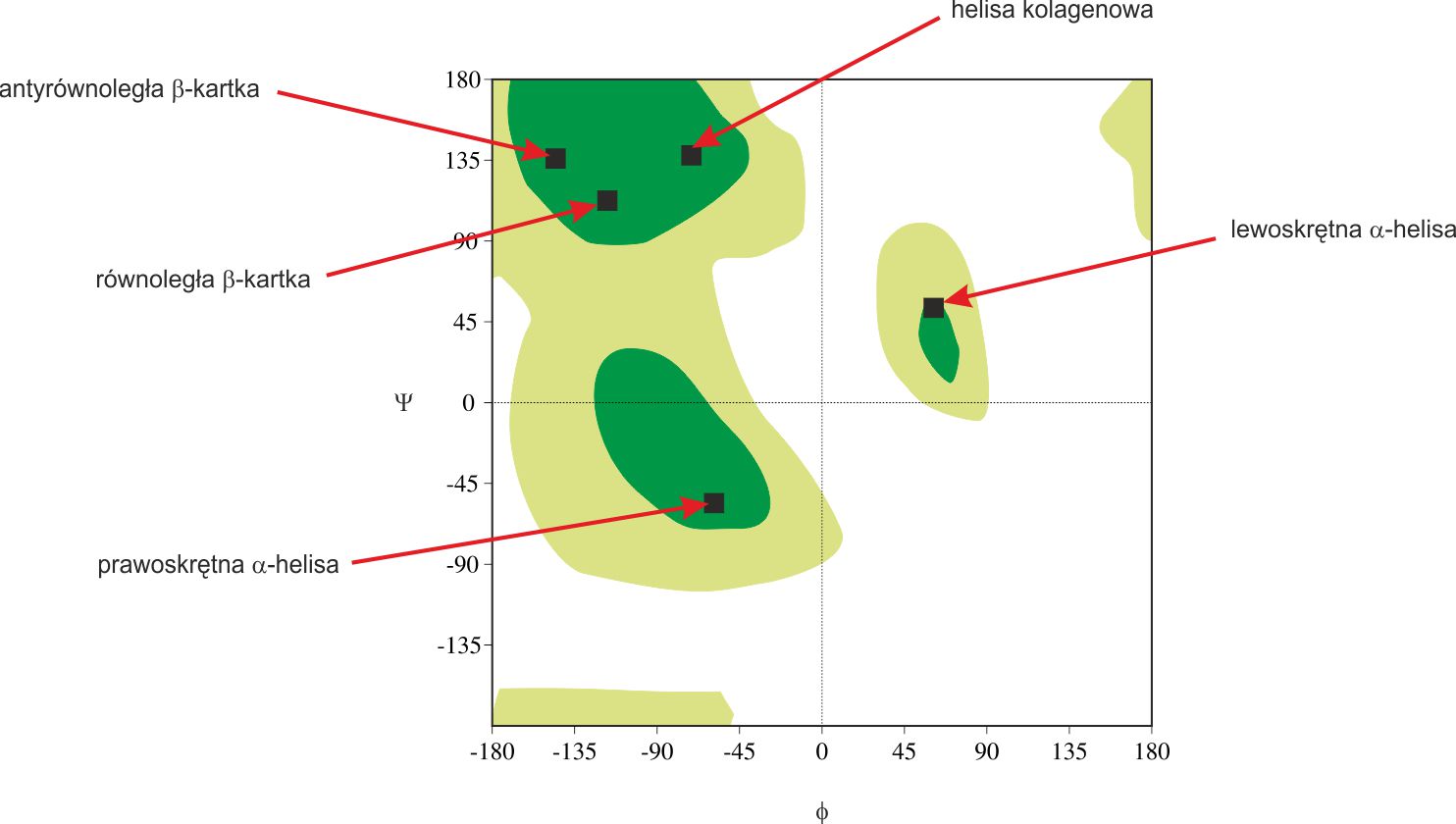
Rysunek 2. Mapa Ramachandrana z zaznaczonymi obszarami uprzywilejowanymi (kolor ciemnozielony) i obszarami dozwolonymi (kolor jasnozielony).
Rysunek 2. Mapa Ramachandrana z zaznaczonymi obszarami uprzywilejowanymi (kolor ciemnozielony) i obszarami dozwolonymi (kolor jasnozielony).
Mapa Ramachandrana jako narzędzie służące do określenia jakości wygenerowanej struktury pokazuje czy konformacja łańcucha głównego jest poprawna. Często są to różnice bardzo subtelne, mające jednak wpływ na całkowitą energię cząsteczki. Otrzymana w wyniku obliczeń struktura może być nie do końca zrelaksowana co przy dużej cząsteczce peptydu lub białka jest niezauważalne gdy ogląda się strukturę pdb. Wiele zawad sterycznych jest wynikiem błędnej konformacji łańcucha głównego biopolimeru. Mapa Ramachandrana jest jednym z najprostszych, najszybszych, a zarazem najbardziej popularnym narzędziem służącym do oceny jakości otrzymanego modelu peptydu lub białka. Na rysunku 3 pokazany jest przykład zawady sterycznej, gdzie dwa atomy tlenu znajdują się zbyt blisko siebie.
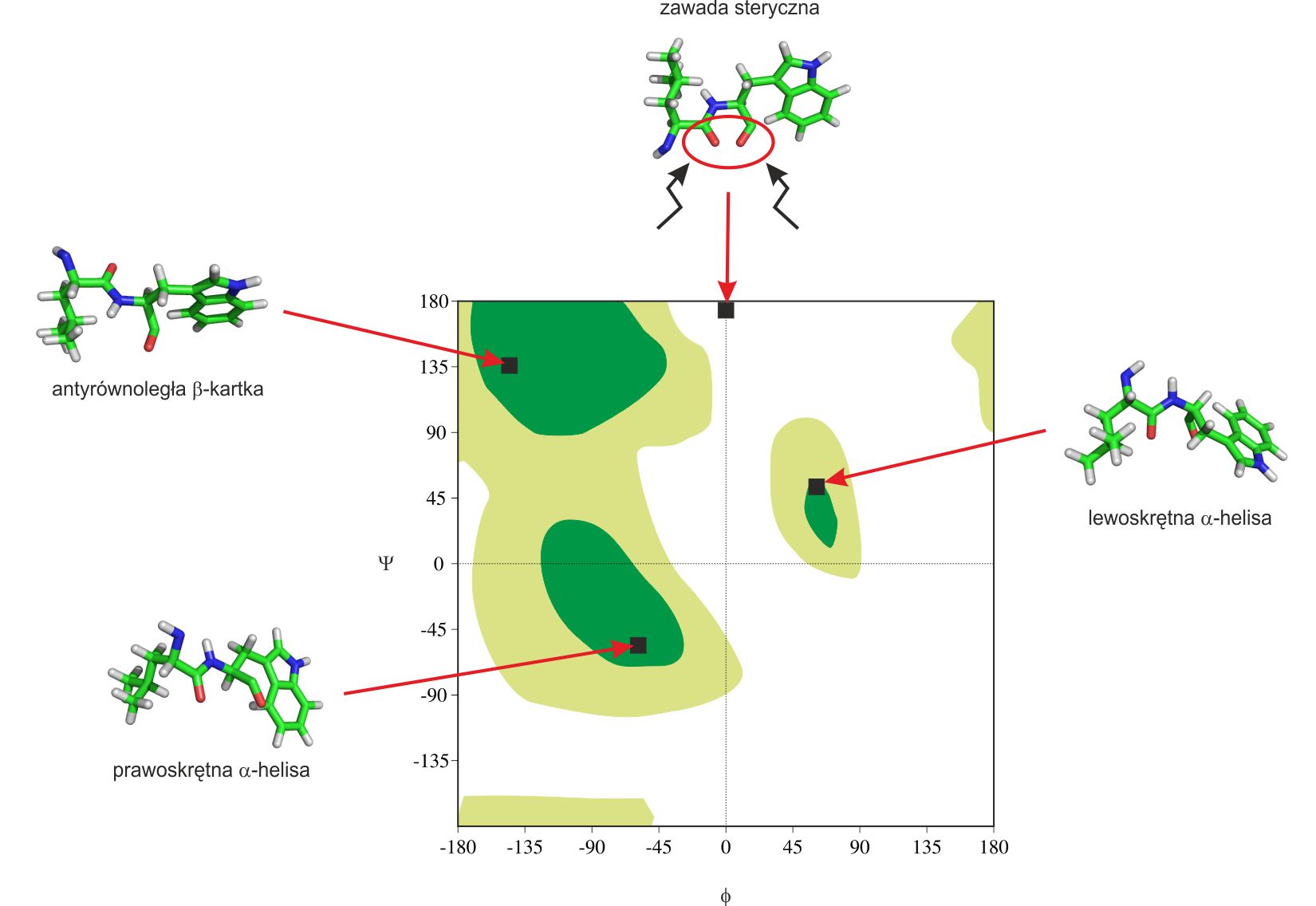
Rysunek 3. Mapa Ramachandrana z zaznaczonymi elementami struktury drugorzędowej oraz przykładem zawady sterycznej.
W konsekwencji cały układ nie jest również uprzywilejowany energetycznie. Dla porównania na rysunku tym znajdują się również poprawnie zbudowane elementy struktur drugorzędowych.
Rysunek 3. Mapa Ramachandrana z zaznaczonymi elementami struktury drugorzędowej oraz przykładem zawady sterycznej.
W konsekwencji cały układ nie jest również uprzywilejowany energetycznie. Dla porównania na rysunku tym znajdują się również poprawnie zbudowane elementy struktur drugorzędowych.
Narzędzia do tworzenia mapy Ramachandrana dostępne są jako darmowe produkty. Godny uwagi jest program ProCheck [2], który tak naprawdę spopularyzował użycie tego wykresu do analizy generowanych struktur. Podejście zastosowane w tym oprogramowaniu dzieli cały wykres na cztery typy obszarów: najbardziej korzystne, dodatkowe dozwolone, dopuszczalne, niedozwolone – rysunek 4.

Rysunek 4. Mapa Ramachandrana wygenerowana w programie ProCheck. Kolorem czerwonym oznaczone są obszary najbardziej korzystne (A, B, L), kolorem żółtym – dodatkowe obszary dozwolone (a, b, l, p), kolorem bladożółtym – obszary dopuszczalne (~a, ~b, ~l, ~p), a kolorem białym – obszary niedozwolone.
Zazwyczaj dobry model peptydu lub białka powinien zawierać 90% wartości kątów ϕ i ψ w obszarze najbardziej uprzywilejowanym. Rozwinięcie mapy Ramachandrana przez autorów Prochecka powoduje, że zapis, iż ponad 80% reszt aminokwasowych znajduje się w obszarze dozwolonym jest nie do końca precyzyjny. Tylko świadome użycie tego narzędzia prowadzi do uzyskania poprawnego modelu. Reszty aminokwasowe, których kąty znajdują się w dodatkowych, dozwolonych obszarach można poddać dodatkowej relaksacji w polu siłowym rozluźniając nieco całą strukturę. Dodatkową zaletą tego pakietu jest możliwość wykonania pełnej statystyki otrzymanej struktury łącznie z procentową zawartością struktur drugorzędowych. Minusem Prochecka może być jedynie fakt, że jest on dostępny jedynie pod system Linux i Unix.
Rysunek 4. Mapa Ramachandrana wygenerowana w programie ProCheck. Kolorem czerwonym oznaczone są obszary najbardziej korzystne (A, B, L), kolorem żółtym – dodatkowe obszary dozwolone (a, b, l, p), kolorem bladożółtym – obszary dopuszczalne (~a, ~b, ~l, ~p), a kolorem białym – obszary niedozwolone.
Zazwyczaj dobry model peptydu lub białka powinien zawierać 90% wartości kątów ϕ i ψ w obszarze najbardziej uprzywilejowanym. Rozwinięcie mapy Ramachandrana przez autorów Prochecka powoduje, że zapis, iż ponad 80% reszt aminokwasowych znajduje się w obszarze dozwolonym jest nie do końca precyzyjny. Tylko świadome użycie tego narzędzia prowadzi do uzyskania poprawnego modelu. Reszty aminokwasowe, których kąty znajdują się w dodatkowych, dozwolonych obszarach można poddać dodatkowej relaksacji w polu siłowym rozluźniając nieco całą strukturę. Dodatkową zaletą tego pakietu jest możliwość wykonania pełnej statystyki otrzymanej struktury łącznie z procentową zawartością struktur drugorzędowych. Minusem Prochecka może być jedynie fakt, że jest on dostępny jedynie pod system Linux i Unix.
Innym narzędziem ogólnodostępnym, pracującym zarówno pod systemem Linux jak i Windows jest Swiss-PdbViewer [3]. Jest to program przeznaczony do wizualizacji cząsteczek, w którym zawarty jest moduł, umożliwiający wygenerowanie mapy Ramachandrana dla oglądanego modelu.
Na poniższych rysunkach przedstawione zostały przykładowe mapy przygotowane w programie Swiss-PdbViewer.
Prezentowane modele pochodzą z bazy struktur PDB (Protein Data Bank) dostępnej pod adresem www.pdb.org. Na rysunku 5 przedstawiono strukturę mioglobiny [4] i wygenerowaną dla niej mapę Ramachandrana. Jak widać większość reszt aminokwasowych znajduje się w obszarze charakterystycznym dla α-helisy. Podobna sytuacja ma miejsce w przypadku rodopsyny wołowej – receptora transbłonowego [5] – rysunek 6. Na rysunku 7 z kolei przedstawiona jest struktura białka VAPB [6], zawierającego motyw klucza greckiego złożonego z rozciągniętych struktur β.
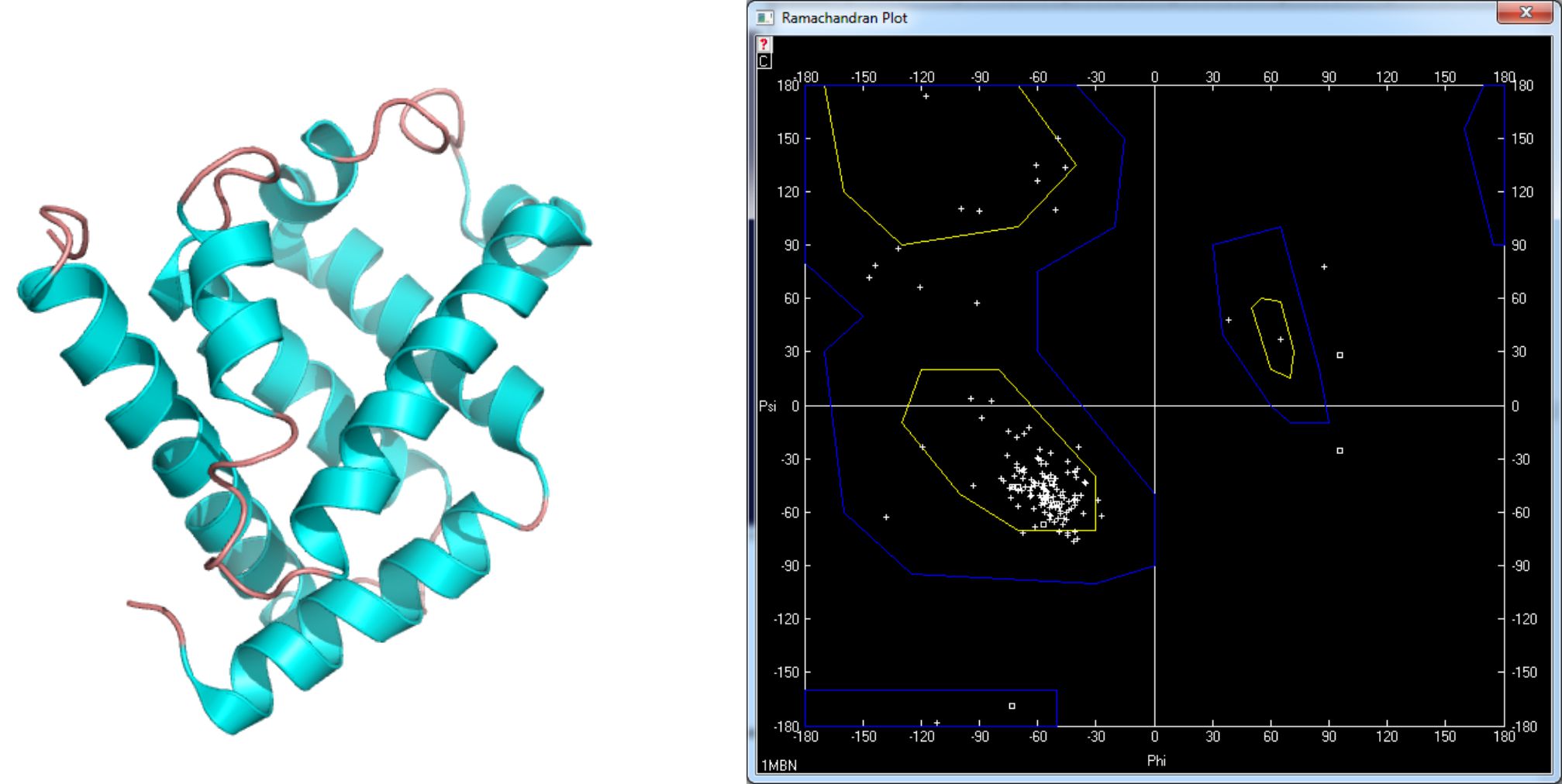
Rysunek 5. Model mioglobiny [1mbn] i wygenerowana mapa Ramachandrana.
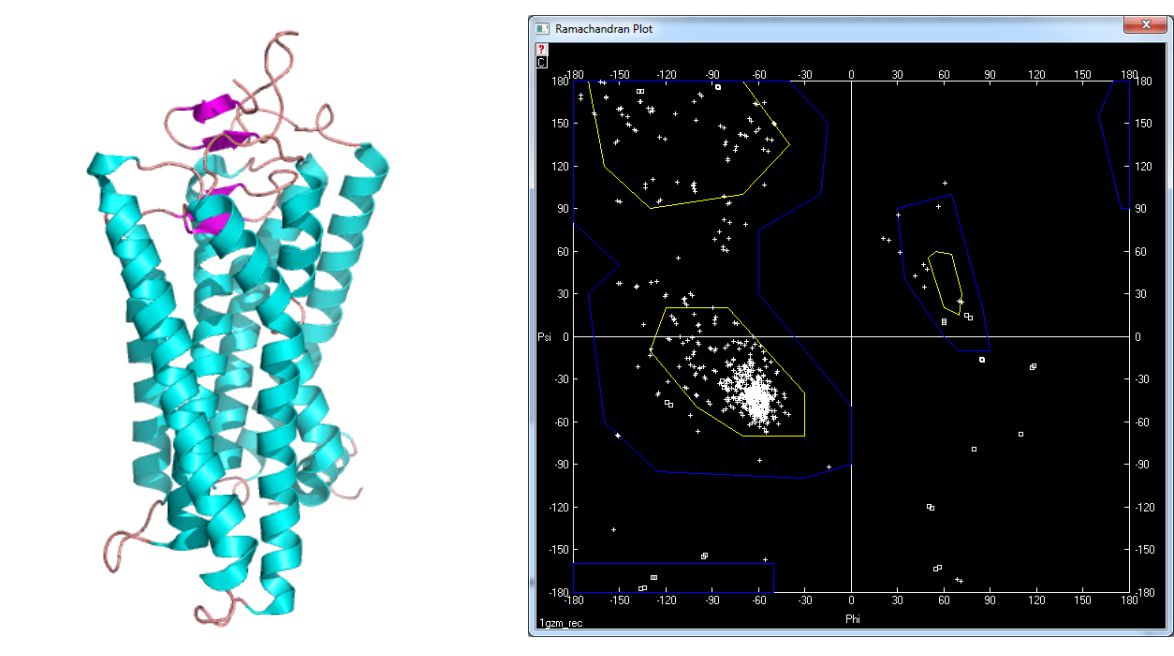
Rysunek 6. Model rodopsyny wołowej [1gzm] i wygenerowana mapa Ramachandrana.
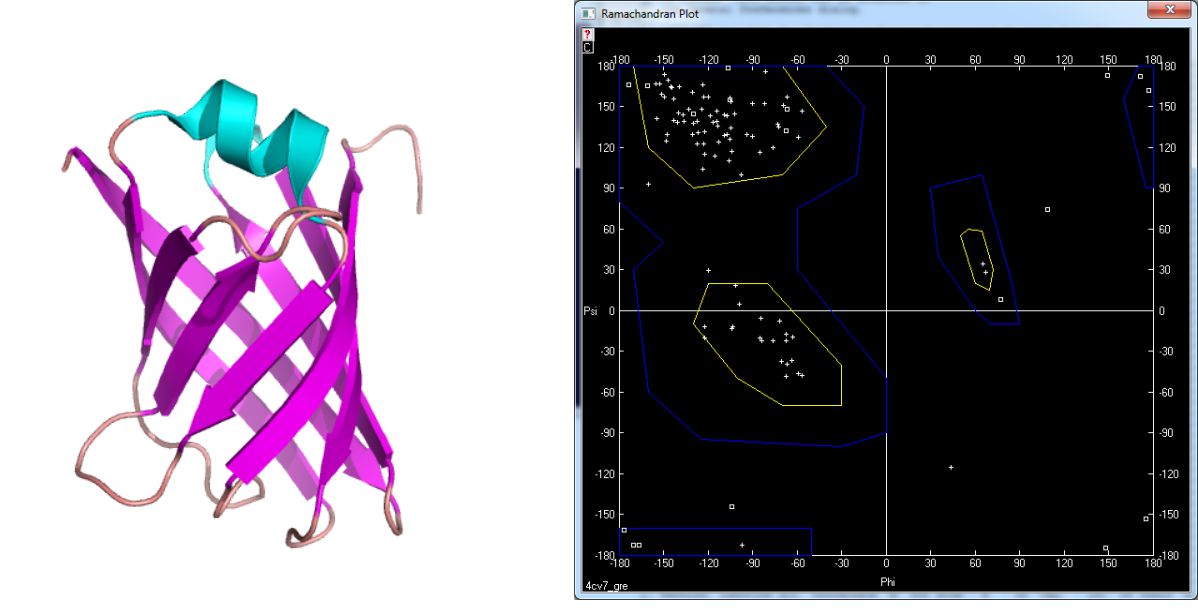
Rysunek 7. Model białka VAPB [4cv7] i wygenerowana mapa Ramachandrana.
Literatura:
[1] G.N. Ramachandran, C. Ramakrishnan, V. Sasisekharan, Stereochemistry of polypeptide chain configurations, J. Mol. Biol., 7, 95 (1963).
[2] R.A. Laskowski, M.W. MacArthur, D.S. Moss, J.M. Thornton, PROCHECK - a program to check the stereochemical quality of protein structures, J. Appl. Cryst., 26,
283, (1993).
[3] N. Guex, M.C. Peitsch, SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling, Electrophoresis, 18, 2714 (1997).
[4] H.C. Watson, The Stereochemistry of the Protein Myoglobin, Prog. Stereochem., 4, 299 (1969).
[5] J. Li, P. Edwards, M. Burghammer, C. Villa, G.F.X. Schertler, Structure of Bovine Rhodopsin in a Trigonal Crystal Form, J. Mol. Biol., 343, 1409 (2004).
[6] C. Geerds, J. Wohlmann, A. Haas, H.H. Niemann, Structure of Rhodococcus Equi Virulence-Associated Protein B (VAPB) Reveals an Eight-Stranded Antiparallel [Beta]-Barrel Consisting of Two Greek-Key Motifs, Acta Crystallogr., Sect. F, 70, 866 (2014).
Rysunek 5. Model mioglobiny [1mbn] i wygenerowana mapa Ramachandrana.
Rysunek 6. Model rodopsyny wołowej [1gzm] i wygenerowana mapa Ramachandrana.
Rysunek 7. Model białka VAPB [4cv7] i wygenerowana mapa Ramachandrana.
Literatura:
[1] G.N. Ramachandran, C. Ramakrishnan, V. Sasisekharan, Stereochemistry of polypeptide chain configurations, J. Mol. Biol., 7, 95 (1963).
[2] R.A. Laskowski, M.W. MacArthur, D.S. Moss, J.M. Thornton, PROCHECK - a program to check the stereochemical quality of protein structures, J. Appl. Cryst., 26,
283, (1993).
[3] N. Guex, M.C. Peitsch, SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling, Electrophoresis, 18, 2714 (1997).
[4] H.C. Watson, The Stereochemistry of the Protein Myoglobin, Prog. Stereochem., 4, 299 (1969).
[5] J. Li, P. Edwards, M. Burghammer, C. Villa, G.F.X. Schertler, Structure of Bovine Rhodopsin in a Trigonal Crystal Form, J. Mol. Biol., 343, 1409 (2004).
[6] C. Geerds, J. Wohlmann, A. Haas, H.H. Niemann, Structure of Rhodococcus Equi Virulence-Associated Protein B (VAPB) Reveals an Eight-Stranded Antiparallel [Beta]-Barrel Consisting of Two Greek-Key Motifs, Acta Crystallogr., Sect. F, 70, 866 (2014).


