


Emilia A. Lubecka
Wydział Chemii Uniwersytetu Gdańskiego
strony wersji drukowanej: 26-34

strony wersji drukowanej: 26-34
Podczas poszukiwania efektywniejszych leków syntezuje się setki, a nawet tysiące nowych związków chemicznych, a następnie analizuje się ich aktywność. Jest to bardzo kosztowny i czasochłonny proces. Obecnie chemia to nie tylko praca w laboratorium nad skomplikowaną aparaturą i z drogimi a często nawet szkodliwymi odczynnikami chemicznymi. Nowoczesne projektowanie leków tzw. „drug-design”, gdzie z powodzeniem wykorzystywana jest chemia teoretyczna, obejmuje również badania nad zależnością struktura-aktywność. Podejście to prowadzi do wyselekcjonowania potencjalnie najlepszych kandydatów na nowy lek - ograniczenia puli rozważanych modyfikacji strukturalnych. Efektem tego jest redukcja kosztów i przyspieszenie w poszukiwaniu bardziej aktywnych i/lub selektywnych leków o oczekiwanych właściwościach fizyko-chemicznych.
Struktura aktywna cząsteczki w roztworze może zostać określona przy zastosowaniu spektroskopii magnetycznego rezonansu jądrowego (NMR). Do badań konformacyjnych zastosowanie znajdują także metody chemii obliczeniowej.
Spektroskopia NMR
Spektroskopia magnetycznego rezonansu jądrowego (ang. Nuclear Magnetic Resonance, NMR) jest jedyną metodą spektralną, która umożliwia pełne poznanie struktury przestrzennej na poziomie atomowym oraz określenie dynamiki badanych biomolekuł w roztworze. W wyniku wprowadzenia technik wielowymiarowego NMR oraz nowych metod obliczeniowych, można obecnie ustalać struktury przestrzenne z dokładnością porównywalną do wyników otrzymywanych na drodze badań rentgenowskich. Ponadto, stosując odpowiednie roztwory, można zapewnić in vitro środowisko zbliżone do warunków natywnych w komórce. Z powyższych względów spektroskopia NMR znajduje szerokie zastosowanie m.in. w monitorowaniu przebiegu procesów biochemicznych; określaniu fragmentów biomolekuł kluczowych dla ich aktywności oraz poszukiwaniu nowych, aktywniejszych leków. Znalazła ona zastosowanie w wielu różnych dziedzinach: od określania struktury chemicznej materii do obrazowania żywych tkanek i całych organizmów.
Podstawy spektroskopii NMR
Istotę spektroskopii NMR stanowi rejestracja sygnałów powstałych wskutek absorbcji promieniowania elektromagnetycznego o częstościach radiowych przez zbiór jąder umieszczonych w polu magnetycznym. Jądra te muszą być magnetycznie czynne, tj. obdarzone spinami i związanymi z nimi momentami magnetycznymi. Najczęściej wykorzystywane są następujące izotopy: 1H, 13C, 15N, 31P oraz 19F. Częstość promieniowania zaabsorbowanego przez dane jądro atomowe jest ściśle zależna od jego typu oraz rozkładu gęstości elektronowej wokół niego – a tym samym od struktury cząsteczki. W efekcie tego, sygnały pochodzące od poszczególnych jąder atomowych badanych cząsteczek charakteryzują się określonym położeniem na widmie. Intensywności tych sygnałów są wprost proporcjonalne do ilości jąder atomowych danego typu. Do określenia położenia sygnału na widmie NMR stosuje się bezwymiarową wielkość zwaną przesunięciem chemicznym δ, której jednostką jest ppm (ang. parts per milion, części na milion). Wyznacza się ją względem przesunięcia chemicznego jąder substancji wzorcowej, w spektroskopii 1H oraz 13C NMR zazwyczaj jest to tetrametylosilan (TMS).
Największe znaczenie spośród metod NMR zyskał magnetyczny rezonans protonowy (1H), ze względu na to, że widoczny w spektroskopii NMR proton jest najbardziej rozpowszechnionym (99,98%) izotopem wodoru, co ułatwia rejestrację widm 1H NMR. Dużą popularnością cieszy się również spektroskopia NMR atomów węgla (13C), azotu (15N) oraz fosforu (31P).
W zależności od tego co jest przedmiotem badań stosowane są różne techniki NMR. Natomiast podstawowe dane uzyskiwane z widm NMR, na podstawie których dokonuje się analizy strukturalnej biomolekuł, są zawsze te same: przesunięcia chemiczne; stałe sprzężenia oraz wielkości jądrowego efektu Overhausera (tzw. kontakty NOE).
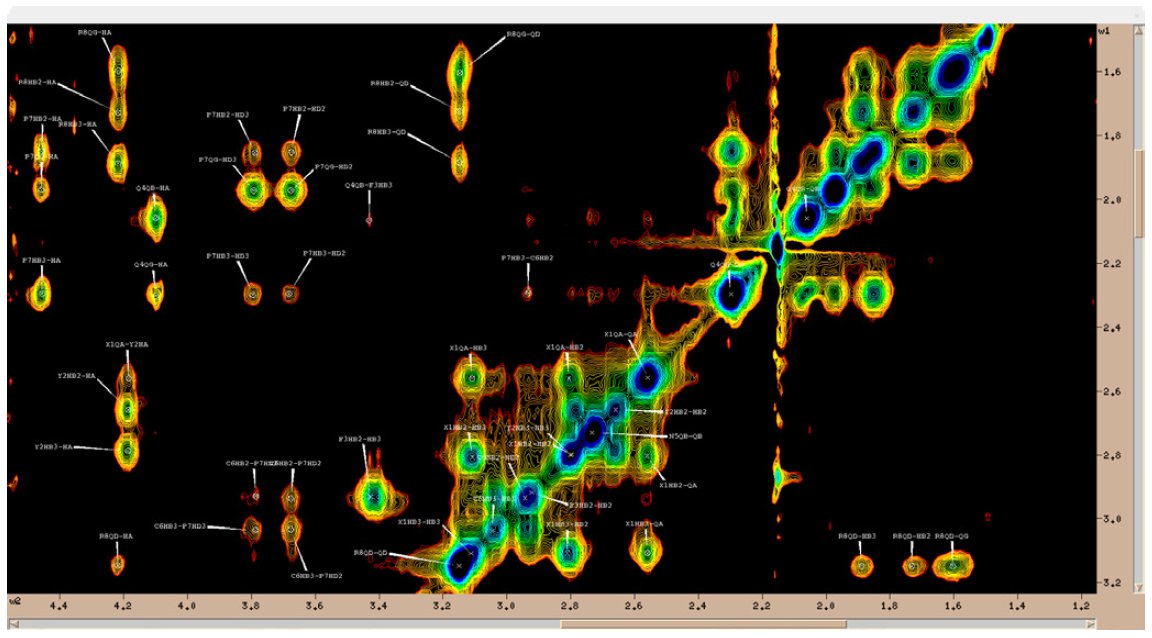
Rys. 1. Przykładowe dwuwymiarowe widmo NMR - widmo NOESY desmopresyny.
Analiza peptydów
W celu przeanalizowania peptydów i niewielkich białek stosuje się klasyczne techniki NMR, opracowane w latach 80-tych XX wieku przez laureata Nagrody Nobla Kurta Wüthricha. Niewielka liczba reszt aminokwasowych - a tym samym niewiele sygnałów na widmach NMR tych związków - sprawia, że nie obserwuje się nakładania sygnałów lub nakładanie to występuje w niewielkim zakresie, z powodzeniem więc można przypisać wystarczającą do analizy strukturalnej liczbę sygnałów bazując na widmach jedno- (1D) oraz dwu- (2D) wymiarowych.
Analizowanie widm NMR peptydów składa się z następujących etapów:
analiza sekwencyjna oraz identyfikacja linii rezonansowych poszczególnych reszt aminokwasowych - uzyskanie informacji o przesunięciach chemicznych atomów wodorów dla poszczególnych reszt aminokwasowych w sekwencji;
uzyskanie informacji o odległościach międzyprotonowych w badanej cząsteczce;
uzyskanie informacji dodatkowych, np.: odczytanie stałych sprzężenia, wyznaczenie współczynników temperaturowych, określenie wartości przesunięć chemicznych węgli itp.
Ogólny schemat procedury oraz techniki NMR wykorzystywane w analizie peptydów przedstawiono na Rysunku 2.
RYS 2
Pewne przesłanki na temat struktury drugorzędowej danych fragmentów peptydów można uzyskać bezpośrednio na podstawie obserwacji kontaktów NOE oraz analizy wartości stałych sprzężenia. Jednakże, do uzyskania pełnego obrazu struktury przestrzennej niezbędne są metody obliczeniowe uwzględniające dane NMR, np. modelowanie molekularne.
Analiza białek
Klasyczne techniki NMR, omówione powyżej, nie sprawdzają się w przypadku dużych biomolekuł, ze względu na nakładanie się licznych sygnałów na widmach, co uniemożliwia ich jednoznaczną identyfikację. Dla białek o masie powyżej 10 kDa, stosuje się widma trój- (3D), cztero- (4D) a nawet pięcio- (5D) czy siedmiowymiarowe (7D), co poprawia rozdzielczość i znacząco zwiększa prawdopodobieństwo rozseparowania poszczególnych sygnałów. Wzrost liczby wymiarów widm NMR, poza zwiększeniem liczby korelowanych jąder (a tym samym większą ilością otrzymywanych danych), niesie ze sobą także znaczny wzrost czasu/kosztu pomiaru. Problem ten został częściowo rozwiązany przez wprowadzenie w ostatnich latach nowych metod transformacji widm. Przykładowo transformacji Fouriera z losowym próbkowaniem (ang. random sampling) przestrzeni czasów ewolucji (ang. evolution time space). Wielowymiarowe techniki NMR wymagają również próbek znakowanych izotopowo – co dodatkowo zwiększa koszty.
Na analizę widm NMR białek, ogólnie ujmując, składają się następujące etapy: analiza sekwencyjna, przypisanie łańcuchów bocznych oraz wyznaczenie odległości międzyprotonowych. Poniżej na rysunku 3 przedstawiono popularniejsze techniki NMR wykorzystywane w badaniach białek w roztworze.
RYS 3
Strukturę przestrzenną białek można wyznaczyć przy użyciu bardzo wielu programów. Część z tych programów bazuje na odległościach pomiędzy protonami (uzyskanymi z kontaktów NOE) np. program CYANA. Inne natomiast już na podstawie sekwencji i zbioru przesunięć chemicznych potrafią wstępnie określić strukturę drugorzędową białka, np. program TALOS+ czy CS23D (ang. Chemical Shift to 3D structure). Do uzyskania pełnej struktury przestrzennej, tak jak w przypadku peptydów, stosuje się metody chemii obliczeniowej.
Metody obliczeniowe
Poznanie struktury peptydu czy białka jest konieczne do zrozumienia jego właściwości fizycznych, chemicznych a także biologicznych. Dane uzyskane z eksperymentu często nie są wystarczające i wymagają weryfikacji metodami obliczeniowymi.
Tworzenie modelu
Przeprowadzanie symulacji komputerowych zawsze należy poprzedzić stworzeniem modelu układu, który chcemy analizować. Dostępne programy posiadają szerokie bazy danych związków chemicznych, które możemy wykorzystać. Długości wiązań, wartości kątów walencyjnych i torsyjnych, ładunki cząstkowe oraz inne parametry charakterystyczne dla budowy poszczególnych związków chemicznych zostały opracowane na podstawie danych eksperymentalnych. W przypadku gdy nie znajdziemy jakiejś substancji, możemy sami dokonać jej parametryzacji i dodać ją do tworzonego przez nas modelu. Możemy stworzyć model samego peptydu, cukru czy wielkocząsteczkowego białka lub większego układu np. biomolekuła - rozpuszczalnik.
Badaniom poddaje się także znacznie większe i bardziej skomplikowane układy, jak np. peptydy czy białka w obecności błony komórkowej, gdyż błony biologiczne odgrywają istotną rolę w stabilizacji struktury oraz w znacznej mierze determinują funkcje znajdujących się w nich białek. Ponadto hormony peptydowe prawdopodobnie wiążą się niespecyficznie z błoną lipidową, przybierając orientację oraz konformację umożliwiającą związanie się z receptorami błonowymi. W związku z modelem dwuetapowego transportu liganda do receptora, istotne jest znalezienie konformacji peptydu w środowisku imitującym błonę komórkową. Ze względu na powyższe, badania konformacyjne często przeprowadza się w układach imitujących środowisko błony komórkowej (Rys. 4). Nie stanowi to problemu dla chemii obliczeniowej. Możemy stworzyć model bardzo skomplikowanego układu i go przeanalizować zanim zdecydujemy się na zakup drogich odczynników chemicznych czy zaplanujemy kolejne eksperymenty.
RYS 4
do receptora, istotne jest znalezienie konformacji peptydu w środowisku imitującym błonę komórkową. Ze względu na powyższe, badania konformacyjne często przeprowadza się w układach imitujących środowisko błony komórkowej (Rys. 4). Nie stanowi to problemu dla chemii obliczeniowej. Możemy stworzyć model bardzo skomplikowanego układu i go przeanalizować zanim zdecydujemy się na zakup drogich odczynników chemicznych czy zaplanujemy kolejne eksperymenty.
Dynamika molekularna
Metoda dynamiki molekularnej (ang. Molecular Dynamics, MD) służy do badania ewolucji czasowej układu molekularnego złożonego z oddziałujących ze sobą w pewnej objętości cząstek. Współczesna chemia obliczeniowa dostarcza wiele pól siłowych umożliwiających analizę zbioru cząsteczek w czasie, takich jak np.: AMBER (ang. Assisted Model Building with Energy Refinement), CHARMM (ang. Chemistry at Harvard Molecular Mechanics) czy ECEPP (ang. Empirical Conformational Energy Program for Peptides). Opisują one hiperpowierzchnię energii potencjalnej układu jako sumę energii oddziaływań pomiędzy poszczególnymi atomami w funkcji współrzędnych tych atomów. Głównym założeniem pól siłowych jest to, że wszystkie oddziaływania pomiędzy atomami są addytywne i niezależne. Pozwala to na przybliżenie energii potencjalnej układu jako sumy energii oddziaływań pomiędzy wszystkimi atomami.
Symulację dynamiki molekularnej przeprowadza się zazwyczaj w dwóch etapach. Pierwszy etap służy ustabilizowaniu układu - uzyskania najbardziej prawdopodobnej konfiguracji badanej cząsteczki w danych warunkach (temperatury, ciśnienia). Uznaje się, że układ osiągnął stan równowagi gdy wartości odpowiednich wielkości termodynamicznych wahają się wokół wartości średnich, które są stałe w czasie. Drugi etap MD polega na zbieraniu danych w różnych warunkach pomiaru. Finalnie otrzymujemy zbiór konformacji, które ze względu na obecność naprężeń atomowych poddaje się następnie minimalizacji energii. Głównym celem minimalizacji jest likwidacja tych naprężeń oraz optymalizacja geometrii badanych układów. Minimalizacja wyszukuje takie położenie atomów, które będzie reprezentować minimum energii potencjalnej systemu. Strukturę przykładowego białka po symulacji MD zamieszczono na rysunku 5.
RYS 5
Określając geometrię badanej cząsteczki metodą MD możemy wykorzystać dane NMR nakładając więzy strukturalne ograniczające ruch danych atomów podczas symulacji. Bardzo często stosuje się procedurę dodawania więzów uśrednionych po danym oknie czasowym (ang. Time-Averaged Restraints, TAV). Takie podejście uwzględnia występowanie badanej cząsteczki w wielu konformacjach będących w równowadze w roztworze, ponieważ odległości międzyatomowe oraz kąty torsyjne są liczone jako wartości średnie w danym oknie czasowym, a nie tylko jako wielkości odpowiadające jednej wybranej konformacji. Dane NMR można wykorzystać również w trakcie analizy i selekcji wynikowych struktur. Najbardziej prawdopodobne są te struktury, które najlepiej odzwierciedlają wyniki eksperymentalne.
Wyznaczenie konformacji badanej biomolekuły na podstawie danych NMR można również sprowadzić do rachunkowego określania struktury poprzez nałożenie więzów na odległości międzyatomowe metodą geometrii odległościowej (ang. Distance Geometry, DG).
Analiza oddziaływań ligand-receptor
Oddziaływania białek z ich ligandami pełnią kluczową rolę w ogromnej ilości procesów biologicznych, a w szczególności w procesach przekazywania sygnału. Dlatego odnalezienie i poprawne zdefiniowanie miejsc na powierzchni lub wewnątrz struktury białka odpowiedzialnych za interakcje z ligandami jest niezwykle istotne. Z tego względu opracowano specjalne programy, które służą do szczegółowej analizy oddziaływań ligand-receptor. Jednym z takich programów „do dokowania” jest program AutoDock.
AutoDock umożliwia zadeklarowanie przestrzeni białka, w której będzie poszukiwał miejsc potencjalnie odpowiedzialnych za wiązanie liganda. Wykorzystuje się w tym celu wprowadzone przez użytkownika dane eksperymentalne dotyczące przybliżonego obszaru wiązania się liganda do receptora. Następnie oblicza mapę powinowactwa atomowego dla każdego atomu w zadeklarowanej przestrzeni dokowania. Mapa ta jest rozpięta na siatce punktów w przestrzeni, w której zostaje umieszczony próbny atom, a potem liczona jest energia oddziaływania pomiędzy białkiem a próbnym atomem.
Program AutoDock generuje dużą liczbę zróżnicowanych konformacyjnie układów sparametryzowanego ligandu w kieszeni wiążącej zamodelowanego receptora. Wybór finalnych konformacji należy do użytkownika i powinien być oparty na wnikliwej analizie dostępnych danych eksperymentalnych. Ponadto ze względu na zastosowane w nim ubogie pole siłowe, otrzymane kompleksy charakteryzują się dużą energią o niewiarygodnym rozkładzie i z tego względu muszą być optymalizowane (np. metodą minimalizacji energii).
W celu określenia zachowania białka receptorowego, jak i identyfikacji reszt receptora potencjalnie zaangażowanych w wiązanie liganda analizuje się zmiany konformacji ligandów podczas symulacji. Otrzymane wyniki mogą zostać wykorzystane m. in. do wskazania potencjalnie istotnych konformerów, mogących być punktem wyjścia w projektowaniu analogicznych związków o znaczeniu farmakologicznym.
W badaniach konformacyjnych dotyczących struktury przestrzennej peptydów i białek istotne jest równoczesne użycie wielu technik badawczych, zarówno doświadczalnych jak i teoretycznych, oraz zestawienie ze sobą uzyskanych różnymi technikami wyników. Pozwala to na dokładne ustalenie struktury przestrzennej biomolekuł i ich dynamiki oraz określenie wpływu geometrii cząsteczki na właściwości biologiczne badanych peptydów czy białek.
Chemia teoretyczna nigdy nie zastąpi eksperymentu, ale może go efektywnie wspomóc ograniczając pulę rozważanych modyfikacji strukturalnych badanych molekuł czy eksperymentów do przeprowadzenia. Może również rzucić światło na przebieg procesów biochemicznych, których na dzień dzisiejszy analiza nie jest możliwa drogą eksperymentalną.
PIŚMIENNICTWO
1. Ejchart A. Spektroskopia magnetycznego rezonansu jądrowego w wyznaczaniu budowy białek. Kosm Probl Nauk Biol. 2004;53:263–270.
2. Forster M. J., Molecular modelling in structural biology. 2002, Micron, 33:365-384.
3. Jacobsen NE. NMR Spectroscopy explained: Simplified Theory, Applications and Examples for Organic Chemistry and Structural Biology. England: Wiley; 2007.
4. Lubecka E.A., Sikorska E. Techniki megnetycznego rezonansu jądrowego w analizie konformacyjnej peptydów i białek . Dokonania Młodych Naukowców. 2014, pod red. M. Kuczera, K. Piech , Creativetime. 5:34-39.
5. Wiórkiewicz-Kuczera J. Metody Pola Sił. Zastosowania w Badaniach Konformacji Układów Biologicznych. Zagadnienia Biofizyki Współczesnej. 1984;9:55–124.


